
Abstract
Chemotherapy-induced peripheral neuropathy (CIPN) is a frequent and dose-limiting side effect of oxaliplatin treatment, yet its molecular mechanisms remain incompletely understood. Calcitonin gene-related peptide alpha (CGRPα, encoded by Calca) is a neuropeptide implicated in several chronic pain conditions and has been proposed to mediate CIPN-related hypersensitivity. Here, we investigated the role of CGRPα in a mouse model of chronic oxaliplatin-induced neuropathy. Mice treated with oxaliplatin over 8 weeks developed cold allodynia and reduced sensory nerve conduction velocity, recapitulating hallmark clinical symptoms of chronic CIPN. However, contrary to expectations, we observed no increase in Calca mRNA expression or protein levels in the dorsal root ganglia (DRG) of male mice and a significant decrease in female mice. The proportion of CGRP-expressing neurons remained unchanged. RNA-seq revealed a two-fold upregulation of Ramp1, a subunit of the CGRP receptor complex. These results suggest that CGRPα signaling may be enhanced not by increased peptide expression, but rather by increased calcium-dependent release from existing neurons and increased CGRP receptor sensitization. This is consistent with known effects of oxaliplatin-induced oxidative stress, which can activate TRPA1 channels and promote calcium-dependent vesicular release of neuropeptides. Although additional validation of this model is needed, our data support a revised rationale of CGRP involvement in CIPN based on sensitization and neuropeptide release, rather than upregulation, and point to TRPA1–CGRP interactions as a potential therapeutic target in oxaliplatin-induced neuropathic pain.
Keywords
Introduction
Oxaliplatin is a third-generation platinum-based chemotherapeutic drug that is widely used to treat various types of cancers, including lung, bladder, cervical, and bowel cancers. 1 Despite its high efficacy, oxaliplatin induces a number of severe side effects,2–4 with chemotherapy-induced peripheral neuropathy (CIPN) being one of the most common and dose-limiting conditions.5–8 Symptoms such as numbness and tingling often lead to dose reductions or discontinuation, limiting treatment intensity and quality of life. Oxaliplatin produces both acute and chronic neuropathy in up to 90% and 70% of patients, respectively.9–11 Notably, the pathophysiological basis of oxaliplatin-induced neuropathy remains poorly understood, and no effective treatments have been developed, particularly for the chronic phase. 12 Despite multiple palliative approaches, their efficacy remains limited, particularly in chronic CIPN. To date, no preventive therapies have shown significant clinical efficacy.13,14
Calcitonin gene-related peptide alpha (CGRPα, encoded by Calca) is a 37-amino-acid peptide primarily found in sensory fibers arising from the dorsal root ganglia (DRG) and trigeminal ganglia. 15 Nerve injuries caused by trauma or inflammation can trigger the release of CGRP and enhance the activity of nociceptors, resulting in thermal and mechanical allodynia.16,17 Numerous studies have revealed that CGRPα serves an important role in pain sensitization and persistence of several neuropathies, including chronic arthritis, endometriosis, and diabetic neuropathy.18–20 Furthermore, blockade of CGRP receptors has proven effective for chronic migraines21,22 with several FDA-approved drugs that function by targeting the CGRP pathway, either by targeting the CGRP peptide itself or the CGRP receptor component, such as RAMP1. 23
The role of CGRP in CIPN, however, has not been fully established. Overexpression of CGRP is a well-documented phenomenon in migraine, contributing significantly to its pathology and serving as a target for therapeutic interventions.24,25 Whether oxaliplatin causes changes in CGRP expression during CIPN was not known. To measure CGRP, we used a mouse model of CIPN through repeated weekly oxaliplatin injections for 8 weeks. The dose of the drug was calculated based on clinical regimens. 26 The progression of CIPN was assessed by cold allodynia and nerve conduction velocity (NCV) measurements. We tested whether chronic oxaliplatin alters Calca expression and CGRPα+ neuronal population in DRG, and based on the results, explored an alternative mechanism behind the pain phenotype.
Methods
Calca denotes the gene encoding CGRPα; Calca mRNA levels and CGRPα protein/peptide levels are distinguished throughout.
Animal models of CIPN
All animal protocols were approved by the Washington University Institutional Animal Studies Committee and were carried out in accordance with NIH guidelines. All mice were housed in a temperature-controlled facility and maintained on a 12:12-h light/dark cycle with free access to water and food.
Thy1-GCaMP6s 4.3 mice
A transgenic 8-month-old female mouse expressing the genetically encoded calcium indicator Thy1-GCaMP6s GP4.3 under the control of a neuronal Thy1 promoter from transgenic mice was received as a gift from Dr. Culver’s lab (Washington University) and sacrificed to extract DRG.
Cold allodynia
Cold allodynia was tested with a hot/cold plate (Ugo Basile) that enables controlled cooling and heating of the plate. Mice were habituated to the room and the plate at room temperature prior to the tests. After the plate was cooled to 4°C, the mice were placed on the plate individually, and the time taken to evoke nociceptive behavior, such as jumping, flinching, hind-paw licking, or lifting, was recorded. The cutoff time was 60 s to avoid tissue damage.
Nerve conduction velocity (NCV)
Sensory NCV was measured on the mouse tail by an automated functional assessment station (FASt System, Red Rock Laboratories (RRL), St. Louis, MO) and its RRL software. Mice were anesthetized with a constant flow of 2% (v/v) isoflurane. During measurement, the body temperature of the mice was maintained at 37°C with a heating pad. The NCV was measured using a battery-powered stimulus isolator (Model 2200, A-M Systems) and an extracellular amplifier (DAM80, WPI Inc.). Stainless steel needle electrodes were used to deliver voltage pulses (0.2 ms duration) distally. Recording electrodes were inserted in the tail 3 cm apart from the stimulating electrodes. Data were analyzed with RRL software, and NCV was calculated by dividing the distance between stimulating and recording electrodes by the latency. The measurement was performed at least 3 times for each mouse.
RT-qPCR
At the end of the study, DRGs were collected and immersed in ice-cold RNAlater (Thermofisher Scientific) before use. Total RNA was extracted from DRG tissue using PureLink RNA Mini Kit (ThermoFisher Scientific) according to the manufacturer’s instructions. First-strand cDNA is synthesized using SuperScript III kit (Invitrogen). Real-time qPCR is run with Quantstudio 3 system (Thermofisher Scientific). Primers for Calca and internal control 18S are as follows: Calca forward – CGCTCACCAGGAAGGCATC, Calca reverse – CATGCCTGGTACAGGAGCAA; 18S forward – AAGTTCCAGCACATTTTGCGAGTA, 18S reverse – TTGGTGAGGTCGATGTCTGCTTTC.
RNA-seq analysis
RNA seq analysis was conducted as we reported earlier. 12 Complete RNA-seq data for DRG are deposited into GEO: GSE286387 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE286387). The data were plotted for each mouse. Fold change (FC) represents the ratio of gene expression in oxaliplatin-treated versus control samples for upregulated genes and − (control/treated) for downregulated genes, showing the magnitude and direction of change. False discovery rate (FDR) adjusted p-values were used to demonstrate statistical significance, with lower FDR values implying higher statistical confidence.
Western blot
Total proteins were extracted from DRGs with radioimmunoprecipitation assay reagent (Cell Signaling Technology) containing 1% phenylmethanesulfonyl fluoride. After denaturation, 20 µg total proteins were loaded to 4%–12% BLOT mini gel and electrically transferred to a nitrocellulose membrane with the iBlot 2 system (Thermofisher Scientific). The membrane was then blocked with 5% non-fat milk in TBS with 1% Tween-20 (TBST) at room temperature for 1 h, followed by incubation with HRP-conjugated mouse anti-CGRP (1:500; Santa Cruz Biotechnology, sc-57053) and HRP-conjugated anti-β-actin (1:1000; Santa Cruz Biotechnology, sc-47778) antibodies in 5% milk TBST overnight at 4°C on a low-speed shaker. The next day, the membrane was washed with TBST and incubated with SuperSignal West Dura chemiluminescent substrate (Thermofisher Scientific) for 1 min. The protein bands are imaged with ChemiDoc (Bio-Rad) system and analyzed with ImageJ software. β-actin ran on the same gel was and was used as an internal control. After transferring the protein to membrane, the membrane was cut based on the protein ladder to stain b-actin and CGRPα separately.
Immunofluorescence
DRGs were fixed in 10% neutral buffered formalin (NBF) overnight at 4°C. For studies with C57BL mice, DRG were embedded in paraffin and sectioned at 5 µm with a microtome. Before antibody staining, paraffin slides were deparaffinized with xylene and rehydrated with 95%, 70%, and 50% ethanol/water solutions, followed by rinsing with DI water. Antigen retrieval was performed by heating in a 1X citric acid buffer with microwave in a pressure cooker for 15 min. Slides were then blocked with a blocking buffer made with TBS containing 0.1% Triton X-100 (TBST) and 10% normal donkey serum (MilliporeSigma, D9663) for 2 h at room temperature (RT). DRG slides were incubated with primary antibodies (anti-CGRPα: 1:750, PA185250, Thermofisher Scientific; anti-NeuN: 1:500, MAB377, MilliporeSigma) diluted in the blocking buffer overnight at 4°C. The following day, sections were washed with TBST 3x10 min and then incubated with secondary antibodies (donkey anti-goat 488, 1:500, A32814, Thermofisher Scientific; donkey anti-mouse 555, 1:500, A32773, Thermofisher Scientific) in TBST for 1 h at RT. The slides were washed 3 × 10 min again in TBST, followed by incubation with DAPI for 10 min at RT. Finally, DRG slides were mounted with Fluoromount mounting medium (MilliporeSigma) and imaged with a fluorescence microscope (Olympus BX51 equipped with an Orca-R2 CCD camera (Hamamatsu).
For immunohistochemistry on CGRPα::TdTomato mice, DRG were embedded in OCT, frozen at −80°C, and sectioned at 10 µm with a cryostat. To retrieve the antigen, slides were immersed in citric acid buffer and incubated at 70°C overnight. The next day, slides were processed as described above with the following antibodies: primary antibody anti-NeuN (1:500, MAB377, Thermofisher Scientific); secondary antibody donkey anti-mouse 488 (1:500, A21202, Thermofisher Scientific).
Measurement of Ca2+ dynamics
DRG were harvested from transgenic mice (8-month-old, female) expressing the genetically encoded calcium indicator Thy1-GCaMPs 4.3 under the control of a neuronal Thy1 promoter. Following dissection, DRG were cultured in a serum-free medium in an incubator maintained at 37°C and 5% CO2 for 3 days. Media was DMEM-based (A4192101, ThermoFisher Scientific) with supplementation of 1% BSA (A7906, Millipore Sigma), 50 μg/mL vitamin C (A4544, MilliporeSigma), 3 g/L extra glucose (G8270, Millipore Sigma), 1% L-glutamine (25030149, ThermoFisher), 1% insulin-transferrin-selenium (51300044, ThermoFisher), and 1% penicillin-streptomycin (15140122, ThermoFisher). Calcium dynamics were monitored using a Bruker Ultima 2Pplus fluorescence microscope equipped with a camera PMT, 25x objective, and a 920 nm excitation source. Baseline fluorescence was recorded for 2–3 min to establish resting calcium levels. Oxaliplatin (final concentration: 150 µM) was then applied directly to the medium, and changes in GCaMP fluorescence intensity (ΔF/F0) were recorded over time to capture neuronal calcium responses. Images were acquired at a frame rate of 0.45 Hz and analyzed using ImageJ v. 1.54 to quantify temporal changes in fluorescence.
Raw fluorescence values (F) were extracted for each time point, and the baseline fluorescence (F0) was defined as the average fluorescence over the first 250 time points (seconds). The relative change in fluorescence (ΔF/F0) was calculated for each time point using the formula:

and the result was multiplied by 100 to obtain percentage values.
Results
Oxaliplatin results in human-like symptoms of peripheral neuropathy in mice
Loss of body weight, cold allodynia, and decrease of NCV are among the most common side effects in patients receiving oxaliplatin. To investigate whether our mouse models developed similar symptoms, we measured their body weight (Figure 1(a)), assessed their response to cold stimuli using a 4°C cold plate assay (Figure 1(b)) and measured NCV (Figure 1(c)). We observed a loss of body weight almost immediately after the first injection of oxaliplatin, whereas cold allodynia became prominent only by week 7. Cold allodynia was evaluated based on latency, measured as the time taken to evoke nociceptive behaviors such as jumping, flinching, and hind paw licking. We measured the sensory NCV on the mouse tail as it offers easier access, minimal invasiveness, and has a relatively uniform size and shape compared to other peripheral nerves in mice body. Here, the NCV measurements were used as a functional indicator of peripheral nerve integrity. This method, however, is less effective for detecting small-fiber dysfunction, which underlies pain and sensory disturbances. Oxaliplatin-treated mice demonstrated a significant reduction in NCV: the control group showed an average of 29.58 ± 3.16 m/s, compared to 23.04 ± 1.32 m/s in the treated group (Figure 1(c)). These results are consistent with findings in humans.28,29 A reduction in NCV suggests potential demyelination and axonal dysfunction. A similar ~20% decrease in NCV in humans would be considered a clinically significant pathology diagnosed as moderate to severe peripheral neuropathy. 30 Overall, these results support that the key clinical features of chronic CIPN in mice are also observed in patients receiving oxaliplatin.

Establishment of a peripheral neuropathy mouse model of CIPN with oxaliplatin. (a) Oxaliplatin led to a decrease in body weight. (b) Mice developed cold allodynia after the 7th repeated administration of oxaliplatin. Week 0 is a baseline measurement. (c) Oxaliplatin-treated mice presented lower NCV at week 8 compared to control animals. N = 5. **p < 0.01
Oxaliplatin impacts on CGRPα levels is sex specific: No change in males, reduction in females
Following the establishment of CIPN symptoms in mice after 8 weeks of treatment, we evaluated the transcriptional and protein levels of CGRPα in the DRG. mRNA expression of CGRP (Figure 2(a)) and CGRPα protein level (Figure 2(b)), determined by qPCR and Western blot, respectively, showed no significant differences between control and oxaliplatin-treated male mice. Consistently, immunofluorescence staining performed to examine the distribution and expression of CGRPα protein in the DRG also did not show differences between the control and treated groups. The fluorescence intensity of CGRPα staining, as well as the percentage of CGRPα immunoreactive neurons (CGRPα+), were comparable between the two groups (Figure 2(c) and (d)).

Oxaliplatin treatment affects the painful phenotype in WT (C57BL/6) male mice but has minimal effect on CGRPα. (a) The mRNA level of Calca in DRG was not altered by oxaliplatin, measured by qPCR. 18S was used as an internal control for quantification. (b) Western blot (see Supplemental Figure S1 for a full blot) and blot quantification of CGRP protein in DRG. The CGRP protein level remained unchanged after oxaliplatin treatment. (c) The percentage of CGRPα+ neurons was calculated using the ratio of CGRPα immunostaining-positive cells to total NeuN-positive cells. No statistically significant differences were detected in (a), (b), or (c). (d) Immunostaining of DRG slices using antibodies against a neuronal marker, NeuN, and CGRPα, DAPI, and merged images of DRG slices. As expected, CGRPα was predominantly expressed in neurons that were NeuN-positive. Scale bar: 100 µm. N = 5.
RNA-seq analysis of DRG from oxaliplatin-treated versus control C57BL/6 mice confirmed the absence of significant changes in Calca expression (Figure 3(a)), but showed a significant increase in the level of Ramp1 expression – a protein that forms a complex with the calcitonin receptor-like receptor (CRLR) to produce the functional CGRP receptor 31 (Figure 3(b)).
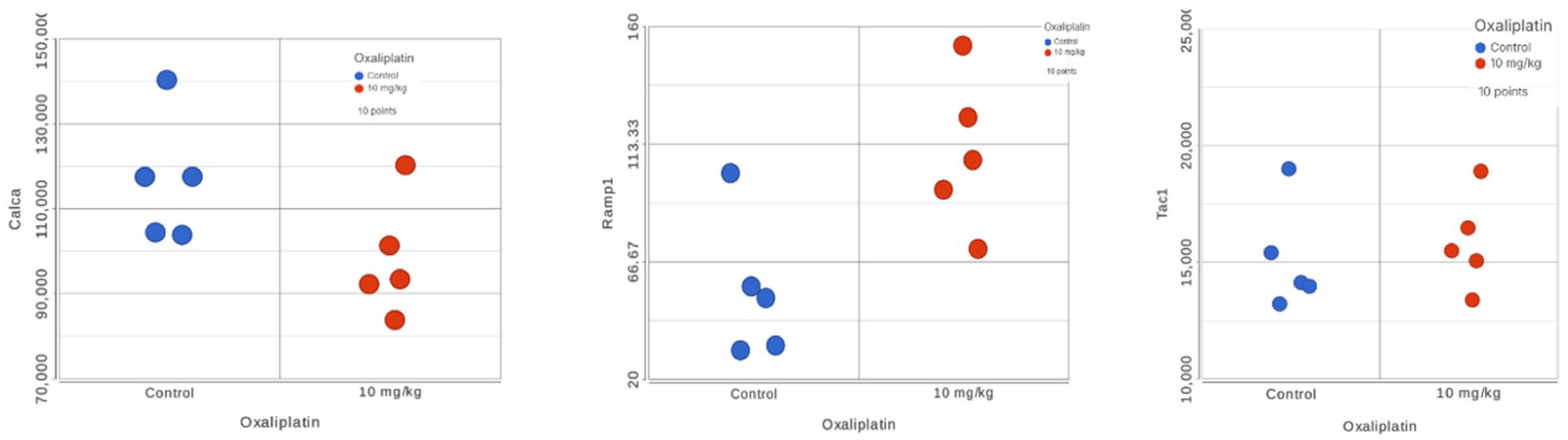
RNA-seq results for the DRG extracted from oxaliplatin treated mice versus control. (a) Calca (FDR = 0.13, FC = −1.19). (b) Ramp1 (FDR = 0.03, FC = +2.00). (c) Tac1 (FDR = 0.80, FC = +1.05). Male mice, N = 5 per group (control vs treated).
Given the sex differences in pain perception and neurobiological responses in humans, 32 we included female mice to ensure a comprehensive evaluation of CIPN development and CGRP expression across sexes. Female mice treated with oxaliplatin exhibited similar changes to male mice, including body weight loss (Figure 4(a)), the onset of cold allodynia beginning at week 7 (Figure 4(b)), and a significant decrease in tail NCV, resulting in a 20% reduction compared to controls (Figure 4(c)).

Oxaliplatin treatment affects CGRP in the WT (C57BL/6) female mice. (a) Oxaliplatin induced significant weight loss in female mice. (b) Development of cold allodynia was observed in oxaliplatin-treated female mice at week 7. (c) Oxaliplatin resulted in a decrease in sensory nerve conduction velocity. (d) The mRNA level of Calca in DRG was lower in oxaliplatin-administered female mice measured by qPCR. 18S was used as an internal control for quantification. (e) Western blot (see Supplemental Figure S2 for a full blot) and blot quantification of CGRP protein in DRG. CGRP protein level decreased after oxaliplatin treatment. (f) The percentage of CGRPα+ neurons was calculated using the ratio of CGRP immunostaining-positive cells to total NeuN-positive cells. (g) Immunostaining of NeuN, CGRPα, DAPI, and merged images of DRG slices. No significant difference was observed in the level of CGRPα+ neurons between the control and oxaliplatin groups. Scale bar: 100 µm. N = 5–8. *p < 0.05, **p < 0.01,****p < 0.0001.
The results from qPCR and Western blot analysis, in contrast, showed a significant decrease in Calca mRNA expression (Figure 4(d)) and CGRP protein level (Figure 4(e)). Immunostaining confirmed CGRPα+ neurons in DRG; however, the percentage of CGRPα+ neurons did not differ significantly (Figure 4(f) and (g)).
Treatment with oxaliplatin does not lead to the preferential loss of CGRPα+ neurons
The unexpected decrease in CGRPα protein levels in female mice prompted us to investigate whether this reduction was due to a decrease in the total number of neurons, the number of CGRPα+ neurons, or a change in CGRPα expression. Previously, oxaliplatin has been shown to cause selective atrophy of a subpopulation of DRG neurons, particularly those expressing large parvalbumin, without inducing cell loss. 33 Whether oxaliplatin reduces CGRPα+ neurons is not known. To address these questions under our conditions, we utilized female CGRPα::TdTomato reporter mice, which express Cre recombinase specifically in CGRPα-expressing neurons upon tamoxifen induction. Tamoxifen was administered at the end of the study (week 6 after the start of oxaliplatin treatments) to induce Cre-dependent neuronal labeling specifically at the chronic stage of neuropathy. This approach enabled visualization and quantification of CGRPα+ neurons at the end of the treatment. It also minimized the potential confounding effects of tamoxifen-oxaliplatin interaction.
Consistent with C57BL/6 mice, oxaliplatin treatment resulted in body weight loss in the CGRPα::TdTomato mice (Figure 5(a)) and the development of cold allodynia, although the symptoms appeared earlier, by week 5 (Figure 5(b)). As shown in Figure 5(c), the CGRPα+/NeuN ratio were not significantly altered (Figure 5(d) shows representative images), suggesting that oxaliplatin treatment did not result in preferential loss of CGRPα+ neurons.

Oxaliplatin treatment affects the painful phenotype in CGRPα::TdTomato female mice but has minimal effect on the number of CGRPα+ neurons. (a) Oxaliplatin led to the loss of body weight. (b) Like WT (C57BL/6) animals, these CGRPα::TdTomato female mice developed cold allodynia after repeated oxaliplatin injections. (c) The percentage of CGRPα+ neurons calculated using the ratio of TdTomato-positive cells to total NeuN-positive cells shows no significant changes. (d) Immunostaining for NeuN, CGRP, DAPI, and merged images from DRG slices. No significant difference was observed in the level of CGRPα+ neurons between the control and oxaliplatin groups. Scale bar: 100 µm. N = 3.
Treatment with oxaliplatin led to an increase in Ca2+-related fluorescence in the DRG neurons
Live-cell calcium imaging of GCaMP-expressing DRG neurons from a Thy1-GCaMP mouse demonstrated a substantial increase in fluorescence following oxaliplatin application and indicated an increase in the intracellular calcium levels. During the baseline measurements, neuronal fluorescence was stable and low (Figure 6(a)). After the introduction of oxaliplatin, a subset of DRG somata and neurons exhibited relatively rapid fluorescence enhancement (Figure 6(b)), consistent with the presence of calcium from either influx or from release from intracellular stores. Quantitative analysis of the normalized fluorescence signal (ΔF/F0) demonstrated a sharp rise within ~5 min of oxaliplatin application, reaching a plateau of approximately six-fold above baseline during the entire 20-min recording (Figure 6(c)) with a small trend of going down with time. These findings show that acute oxaliplatin application elicits strong Ca2+ responses in cultured DRG neurons.

An increase in fluorescence of DRG extracted from a Thy1-GCaMP mouse before and after contact with oxaliplatin leads to Ca2+ influx within several minutes. (a) A representative image of DRG baseline emission. (b) A representative image of DRG emission after oxaliplatin treatment. (c) ΔF/F0 (%) trace of a region shown in B, where F0 was defined as the mean fluorescence of the first 250 s measurements.
Discussion
The goal of the presented study was to investigate whether chronic oxaliplatin treatment contributes to CIPN via CGRP in the DRG. The DRG explant model enables direct interrogation of neuron–glia–redox pathways within the DRG without confounding systemic variables. While this model preserves native neuronal–satellite glial architecture, it does not fully recapitulate the in vivo microenvironment of CIPN, including interactions with circulating immune cells, systemic cytokines, and vascular components. The DRG explant assay provides an intermediate platform between dissociated cultures and in vivo models, such as our recently published mice models of oxaliplatin induced peripheral neuropathy, 8 and is particularly suited for mechanistic studies and early-stage therapeutic screening.
Our results showed that CGRP expression in DRG, measured at both the mRNA and protein levels, was not increased as CIPN developed in male mice. We detected even lower Calca mRNA and CGRPα expression and protein levels in female mice. The reduction in Calca mRNA and CGRPα protein observed in female mice occurred without a corresponding change in the number of CGRPα+ neurons, indicating the changes were associated with neuropeptide availability rather than neuronal loss. In addition to the decreased CGRP expression in female mice, lowered CGRPα protein levels could also be associated with increased peptide degradation, altered axonal transport, or compensatory modulation of related neuropeptide systems in female mice.
Major downstream proteins of the CGRP pathway showed no significant changes except for Ramp1 upregulation. Ramp1 forms part of the CGRP receptor complex. These findings challenge the commonly accepted view that increased CGRP expression drives CIPN and highlight a sensitization-driven mechanism.
Substance P (SP) is often co-expressed with CGRP in DRG neurons and can, in principle, provide a compensatory mechanism for pain. Notably, the lack of a statistically significant difference in Tac1 expression (Figure 3(c)), a gene that encodes essential peptide neurotransmitters, including SP, Neurokinin A, etc. does not support activation of this compensatory mechanism at the transcript level. Interestingly, a recent paper challenges the whole rationale that CGRP and SP are involved in pain. 34 Although Tac1 transcript levels were unchanged, compensatory neuropeptide signaling may still occur through post-transcriptional or activity-dependent mechanisms that were not assessed.
The release of CGRP from neurons is typically mediated by exocytosis and triggered by an increase in intracellular calcium concentration, which can be mediated by various stimuli such as activation of transient receptor potential (TRP) channels. One such channel is TRPA1 35 that has been implicated in inflammatory and neuropathic pain and is a potential target for nociceptive pain. 36 When activated, TRPA1 can elevate intracellular calcium levels that, in turn, trigger vesicle exocytosis via the action of SNARE proteins and subsequent CGRP release. 37 TRPA1 in DRG neurons has been demonstrated to be oxidized by acute oxaliplatin treatment 38 and such oxidation leads to the sensitization of TRPA1 ion channels and CGRP release.38,39 Indeed, our experiments using DRG from Thy1-GCaMP mice confirmed an increase in intracellular Ca2+ following oxaliplatin application as judged by the increase in fluorescence intensity after drug addition. Thus, oxidized TRPA1 may increase cytoplasmic Ca2+ concentrations, which could facilitate CGRP release.
This oxaliplatin-oxidized TRPA1-cytoplasmic Ca2+ influx-CGRP release model could be potentially behind oxaliplatin-induced cold allodynia, one of the most prominent symptoms observed in patients undergoing oxaliplatin treatment.40,41 Cold hypersensitivity in DRG neurons is known to be mediated by TRPA1 ion channels, which are typically inactive unless triggered by noxious cold.42,43 Oxidative modifications of TRPA1 caused by reactive oxygen species generated by oxaliplatin treatments are expected to sensitize the channels to various stimuli, including cold. This sensitization shifts the temperature threshold required for TRPA1 activation to less cold temperatures,43,44 leading to TRPA1 channel opening, Ca2+ influx45,46 (as we observed), and ultimately the release of CGRP and potentially other neuropeptides through vesicular exocytosis.47,48 Chronic activation of TRPA1 channels can enhance calcium-dependent neuropeptide release from sensory neurons. However, the observed upregulation of the CGRP receptor component RAMP1 might also reflect adaptive sensitization of CGRP-responsive cells rather than a direct result of the increased CGRP availability. Because the cellular localization of CGRP receptors within the DRG is not fully defined, RAMP1 upregulation may occur in non-neuronal populations, such as satellite glial or other supporting cells, where increased RAMP1 expression can be independent of CGRP levels. This interpretation is based on the increased level of Ramp1 expression. The functional CGRP receptor signaling requires heteromerization of the calcitonin receptor with RAMP1 and appropriate trafficking to the plasma membrane. These regulatory steps will require validation at the protein and signaling levels.
Calcium imaging in this study was performed acutely following oxaliplatin exposure and therefore reflects early calcium signaling and excitability changes rather than long-term calcium homeostasis. Chronic CIPN potentially involves sustained dysregulation of intracellular calcium handling, including mitochondrial calcium overload, ER stress, and impaired buffering capacity, which were not directly assessed in the current work. Nevertheless, acute calcium responses provide a sensitive functional readout of early neuronal perturbation and may represent initiating events that trigger downstream events such as mitochondrial dysfunction, oxidative stress, and inflammatory signaling. Future studies combining longitudinal imaging, organelle-specific calcium probes, and in vivo validation will be required to clarify chronic calcium dysregulation in CIPN.
Supporting our proposed mechanism, recent studies have shown that pharmacological inhibition or TRPA1 knockout in mice suppresses cold allodynia in models of CIPN. 49 Interestingly, a recent study demonstrated that Substance P and CGRPα-knockout mice still experience oxaliplatin-induced pain, 34 suggesting that CGRP is not the only mediator of pain and indicates the involvement of additional neuropeptides or downstream signaling pathways contributing to the neuropathic pain from oxaliplatin.
Conclusions
Our study demonstrates that chronic oxaliplatin treatment induces classic symptoms of CIPN in mice, including cold allodynia and reduced nerve conduction velocity, in both sexes. However, we found no evidence of Calca overexpression in the DRG during the development of chronic neuropathy. In fact, the CGRPα protein levels remained unchanged in males and were reduced in females following oxaliplatin treatment. Despite these findings, behavioral symptoms of neuropathy were pronounced, suggesting that increased CGRP expression is not required for the manifestation of oxaliplatin-induced chronic pain. Instead, our findings support an alternative mechanism in which TRPA1 channel sensitization, triggered by oxaliplatin-induced oxidative stress, promotes calcium influx and enhances vesicular CGRP release from pre-existing CGRPα neurons. Moreover, this sensitization-driven pathway is supported in part by upregulation of Ramp1, a key CGRP receptor complex component of the, consistent with increased CGRP pathway sensitivity. These results challenge the prevailing assumption that CGRP upregulation is central to CIPN pathogenesis and instead point to a release-based mechanism.
The proposed TRPA1-calcium-CGRP pathway, however, is currently supported by indirect evidence. Direct functional modulation of TRPA1 or CGRP signaling was not addressed here and will be the focus of future studies. Such studies will be necessary to validate the temporal and causal relationships between TRPA1 activation, calcium dysregulation, and CGRP-mediated nociceptive signaling in chronic CIPN.
While the proposed mechanism requires further validation, targeting this signaling axis may offer a novel therapeutic avenue for treating chronic neuropathic pain associated with oxaliplatin.
Supplemental Material
sj-docx-1-mpx-10.1177_17448069261432028 – Supplemental material for CGRP expression and signaling sensitization in a mouse model of chronic oxaliplatin-induced peripheral neuropathy
Supplemental material, sj-docx-1-mpx-10.1177_17448069261432028 for CGRP expression and signaling sensitization in a mouse model of chronic oxaliplatin-induced peripheral neuropathy by Junwei Du, Leland C Sudlow, Margaret H Johnson, Kanishk Satish, Abraham Villagomez, Hongzhen Hu and Mikhail Y Berezin in Molecular Pain
Footnotes
Acknowledgements
We thank the Siteman Cancer Center (SCC) and the Institute of Clinical and Translational Sciences (ICTS) at Washington University in St. Louis for using the Genome Technology Access Center. The SCC is supported in part by an NCI Cancer Center Support Grant #P30 CA091842, and the ICTS is funded by the National Institutes of Health’s NCATS Clinical and Translational Science Award (CTSA) program grant #UL1 TR002345. We also thank the Washington University Center for Cellular Imaging (WUCCI) for imaging studies. We thank Dr. Yameng Xu for helping with the measurement of NCV, and Dr. Ikbal Sencan-Egilmez, Katie Duncan, and Yitian Zhang for help with the 2P microscope for calcium dynamics. We thank Dr. Joe Culver for providing Thy1-GCaMP6s 4.3 mice.
Author contributions
JD, MJ, KS, and AV conducted experiments, analyzed data, and wrote the experimental part; JD, HH, and MYB designed the experiments; JD, LCS, and MB wrote the manuscript and analyzed data. MB provided funding. All authors authored the manuscript, with editing contributions from all the authors.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MB is the founder and owner of HSpeQ LLC that licensed IDCubePro software from Washington University, and a consultant for Daxor Inc and Sarya LLC. These companies had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results, and did not contribute financially to this work. The other authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: NIH/NCI R01CA208623, R21CA269099, NIH/NINDS R21NS135646, and NIH/NINDS 1R01NS139461 (all MYB).
Ethical considerations
All animal protocols were approved by the Washington University Institutional Animal Studies Committee and were carried out in accordance with NIH guidelines.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.