
Abstract
Purpose:
To investigate evodiamine’s analgesic effects and molecular mechanisms on chemotherapy-induced peripheral neuropathy (CIPN), focusing on the p38/MAPK-TRPV1 signaling axis and macrophage polarization in dorsal root ganglia (DRG).
Methods:
A paclitaxel-induced CIPN rat model was established with behavioral assessments via von Frey and thermal hyperalgesia tests. TRPV1, TRPV4, and inflammatory cytokine expression were analyzed using qRT-PCR, ELISA, and Western blot. Macrophage infiltration and polarization were evaluated by flow cytometry and immunofluorescence. Mechanistic studies utilized macrophage-conditioned media from RAW264.7 cells and clodronate liposome-mediated macrophage depletion to establish causal relationships between macrophage polarization and nociceptive behavior.
Results:
Evodiamine dose-dependently alleviated paclitaxel-induced mechanical and thermal allodynia both acutely and preventively. It selectively inhibited upregulation of TRPV1 without affecting TRPV4 and reduced pro-inflammatory cytokine levels (TNF-α, IL-1β, IL-6, MCP-1) in the DRG. Evodiamine significantly reduced F4/80+ macrophage infiltration and shifted macrophage polarization from a pro-inflammatory M1 phenotype (decreased MCP1, CD86) to an anti-inflammatory M2 phenotype (increased CD163, CD206). Notably, macrophage-conditioned medium experiments revealed that evodiamine indirectly modulates neuronal TRPV1 expression through macrophage-derived factors. Furthermore, evodiamine attenuated paclitaxel-induced p38 MAPK phosphorylation in DRG neurons, with selective p38 MAPK inhibition by SB203580 confirming this pathway’s critical involvement in TRPV1 regulation and pain modulation.
Conclusion:
Evodiamine alleviates CIPN through a novel neuroimmune mechanism involving M2 macrophage polarization and inhibition of the p38/MAPK-TRPV1 axis in DRG neurons. These findings establish macrophage polarization as a key therapeutic target and highlight evodiamine’s potential as a natural therapeutic agent for CIPN management.
Keywords
Introduction
Chemotherapy-induced neuropathic pain (CINP) is a prevalent, severe, and persistent adverse effect of anti-tumor therapies, manifesting as diverse peripheral neuropathy symptoms including numbness, pain, burning sensations, tingling, temperature hypersensitivity, mechanical hyperalgesia, and reduced voluntary activity.1,2 The pathophysiology of CINP primarily involves axonal degeneration-induced neuropathic pain, with significant contributions from oxidative stress, inflammation, and aberrant glial cell activation.3,4 CINP can emerge during chemotherapy administration, occasionally necessitating dose reduction or premature termination of treatment. While symptoms typically attenuate following chemotherapy cessation, some patients experience chronic manifestations persisting for months or years. 5 The incidence of CINP following neurotoxic chemotherapy approximates 68% at 1 month, 60% at 3 months, and 30% at 6 months post-treatment. 6 Despite its substantial impact on patients’ quality of life and treatment outcomes, effective preventive and therapeutic strategies for CINP remain limited.
Evodiamine, a quinolone alkaloid and principal bioactive constituent of Evodia rutaecarpa (Wu-Chu-Yu) fruit, has been utilized in traditional Chinese medicine for conditions including migraines, owing to its cold-dispelling, analgesic, antiemetic, and antidiarrheal properties. 7 This compound exhibits diverse pharmacological activities encompassing cardioprotection, anti-tumor effects, anti-inflammatory actions, anti-obesity properties, anti-allergic capabilities, analgesic effects, anti-ulcer activity, and neuroprotection.8–10 Mechanistically, evodiamine modulates Treg and Th17 cell differentiation while inhibiting inflammatory cytokine production. 11 It interacts with the TRPV1 channel to mitigate peripheral hyperalgesia in murine models, demonstrating significant analgesic efficacy. 12 Furthermore, evodiamine exhibits potential to suppress neuroinflammation through modulation of the AKT/Nrf2/HO-1/NF-κB signaling axis.13–16 Our previous investigations revealed that evodiamine significantly inhibits inflammatory mediator release in the dorsal root ganglia (DRG) of rats with chemotherapy-induced neuropathic pain, thereby ameliorating pain symptomatology. 17 However, the precise molecular mechanisms underlying evodiamine’s analgesic effects warrant further elucidation.
Macrophages represent critical cellular mediators in neuroinflammatory processes and pain signaling pathways.18,19 Based on their activation state, macrophages are categorized into pro-inflammatory M1 and anti-inflammatory M2 phenotypes. M1 macrophages predominantly participate in inflammatory cascades, secreting substantial quantities of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6, which significantly contribute to CINP pathogenesis and progression. 20 Conversely, M2 macrophages primarily facilitate tissue repair and resolve inflammation through secretion of anti-inflammatory mediators such as IL-10 and TGF-β. 21 In CINP models, enhanced macrophage infiltration and M1 activation in the DRG exacerbate inflammatory responses and intensify pain manifestations. 22 Therefore, therapeutic strategies targeting macrophage polarization—particularly promoting M2 activation—may represent an effective approach for CINP management.
Transient receptor potential vanilloid receptor 1 (TRPV1), initially identified as the capsaicin receptor, belongs to the TRP channel vanilloid receptor subfamily. 23 TRPV1 channels exhibit widespread distribution throughout the somatosensory system, particularly within primary afferent neurons, where they integrate diverse nociceptive stimuli and facilitate pain signal transmission from the DRG and spinal cord, contributing significantly to pathological and inflammatory pain states.24–26 In various neuropathic pain models—including chronic sciatic nerve constriction injury, diabetic neuropathy, and CINP—TRPV1 expression is markedly upregulated in ipsilateral DRG neurons.27–29 TRPV1 undergoes sensitization during nociceptive signal transmission through phosphorylation via protein kinase C and cAMP-dependent protein kinase A. Following peripheral sensory fiber damage, enhanced TRPV1 receptor expression promotes cellular calcium influx, triggering release of pro-nociceptive substances such as substance P and calcitonin gene-related peptide, thereby inducing pain, hyperalgesia, and allodynia. 30 In paclitaxel or oxaliplatin-induced CINP animal models, increased DRG TRPV1 expression correlates with mechanical allodynia and hyperalgesia.31–33 Moreover, TRPV1 participates in diverse inflammatory processes within the nervous system, cutaneous tissues, and respiratory tract.34–36 While TRPV1’s involvement in CINP and inflammatory cascades is well-documented, its role in mediating evodiamine’s analgesic effects remains incompletely characterized. TRPV1 may represent a pivotal mechanistic target in CINP pathogenesis and contribute to amplified inflammatory responses, though precise mechanisms require further investigation.
In this study, we evaluated evodiamine’s effects on CINP and elucidated potential molecular mechanisms underlying its therapeutic activity. Our findings demonstrate that evodiamine effectively alleviates paclitaxel-induced nociceptive behaviors in experimental animals. Furthermore, repeated evodiamine administration inhibits paclitaxel-induced TRPV1 upregulation and suppresses release of pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, and MCP-1 in the DRG. These results suggest that evodiamine exerts analgesic effects by inhibiting TRPV1 channel upregulation and attenuating pro-inflammatory cytokine release in the DRG of paclitaxel-induced CINP rats. This mechanistic insight enhances our understanding of evodiamine’s therapeutic activity in chemotherapy-induced neuropathic pain. This investigation provides valuable perspective on evodiamine’s potential therapeutic applications in CINP management and highlights its modulatory effects on TRPV1 signaling and neuroinflammatory processes in the context of neuropathic pain.
Methods
Experimental animals and paclitaxel administration
Six- to eight-week-old male Sprague-Dawley (SD; 200–220 g) were purchased from the GemPharmatech (Nanjing). Sample size for this study was determined based on methods described in previously published references.37–40 The rats were housed individually in temperature-controlled cages at 24 ± 1°C with free access to food and water. They were allowed to acclimate to the experimental conditions for 1 week before the start of the experiment.
Subsequently, a paclitaxel-induced peripheral neuropathy model was established following methods described in previous studies.39,41 This dosing regimen effectively induces typical mechanical allodynia and thermal hyperalgesia in rats, closely resembling neuropathic symptoms observed in clinical chemotherapy. Briefly, paclitaxel was dissolved in 10% DMSO to prepare a 100 mg/ml stock solution, which was then diluted with PBS to a working concentration of 1 mg/ml (w/v) containing 0.1% DMSO. Paclitaxel was administered intraperitoneally to the rats at a dose of 2 mg/kg and a volume of 0.5 ml, twice a week on days 1 and 4, for a total of six injections, achieving a final cumulative dose of 12 mg/kg. The vehicle group received intraperitoneal injections of 0.5 ml PBS buffer containing 0.1% DMSO, while the blank group received an equivalent volume of PBS buffer to exclude solvent effects on the experiment. After paclitaxel injection, the rats were given daily intraperitoneal injections of evodiamine (dissolved in PBS solution containing 0.1% DMSO) at doses of 0.3, 1, or 3 mg/kg. The vehicle group received an equal volume of evodiamine solvent (PBS buffer containing 0.1% DMSO). Behavioral tests were conducted on days 3, 7, 14, and 21 to assess their effects on the rats. The experiments conducted in this study were approved by the Animal Welfare Committee of the Second People’s Hospital of Changzhou Affiliated to Nanjing Medical University.
Intrathecal injection of TRPV1 antagonist
The TRPV1 antagonist capsazepine (MCE, HY-15640) was dissolved in DMSO and then diluted with PBS to the dosing concentration. Rats received intrathecal injections of 30 μg capsazepine via subarachnoid administratio daily, 30 min prior to evodiamine treatment.42,43 The vehicle group received an equal volume of PBS buffer containing 0.1% DMSO.
Mechanical paw withdrawal threshold (PWT) measurement
In the mechanical allodynia (von Frey) test, the rats were placed in plastic cages on a wire mesh rack and allowed to acclimate to the environment before testing. The rats’ sensitivity to mechanical pain was assessed using von Frey nylon filaments by observing their paw withdrawal responses to different filament tensions. The von Frey nylon filaments were used to stimulate the mid-plantar surface of the left hind paw of each rat, starting from 1 g and ranging from 1 to 15 g. The nylon filament was applied to the hind paw until it bent, and it was kept perpendicular to the implanted side of the paw for about 6–8 s. The paw withdrawal threshold of the rats, measured in grams, was the final test result of this procedure.
Thermal paw withdrawal latency (PWL) measurement
In the thermal hyperalgesia test for rats, the paw withdrawal latency of all rats was assessed using a hot plate test to evaluate thermal hyperalgesia. The hot plate device consisted of a 25 cm × 25 cm metal plate placed inside a glass enclosure. Before the test procedure, each rat was placed on the hot plate for 10 min without heating to acclimate. The rat’s hind paws were ensured to gently contact the surface of the plate without applying pressure. The hot plate temperature was set at 54°C, with a cutoff time of 30 s. The time when the rat shook, withdrew, or licked its paw was recorded as the pain threshold. The threshold data were calculated based on the results of three experiments, with each experiment separated by 15 min.
Enzyme linked immunosorbent assay (ELISA)
DRG samples from the ipsilateral L4-L6 segments of each rat in the experimental groups were collected, and the homogenates and supernatants of the samples were promptly collected at low temperature. ELISA kits (R&D Systems, USA) were used to determine the protein levels of TNF-α, IL-1β, IL-6, and MCP-1.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
In the RT-qPCR procedure, total RNA was extracted from the DRG using TRIzol reagent, and the RNA concentration of each sample was measured using a Nanodro2000 UV spectrophotometer. Subsequently, 1 μg of each RNA sample was reverse-transcribed into cDNA, and qPCR experiments were performed using the PrimerScript MTRT Master Mix Kit. The qPCR reaction was carried out according to the instructions of the SYBR® Premix ExTaqTM kit. The reaction included pre-incubation at 95°C for 3 min, followed by 40 cycles of gene fragment amplification at 95°C for 10 s and 58°C for 10 s. Gene expression levels were quantified using the 2-ΔΔCq method, with the expression level of the GAPDH gene as an internal reference. The primer sequences are as follows: Trpv1: F: 5′-TGACAGCGAGTTCAAAGACC-3′, R: 5′-CTGGCATTGACAAACTGCTT-3′; Trpv2: F: 5′-AGACGTGCCTGATGAAGGC-3′, R: 5′-TGCACCAAGCAGTGGGAT-3′; Trpv3: F: 5′-AAGAAGAGTGCACACTTCTTCCTG-3′, R: 5′-TTCATCAGGCAGGTCTTCCC-3′; Trpv4: F: 5′-CAGCAAGATCGAGAACCGCCAT-3′, R: 5′-CGAACTTACGCCACTTGTCCCT3′; GAPDH: F:5′-TGATTCTACCCACGGCAAGTT-3′, R:5′-TGATGGGTTTCCCATTGATGA-3′.
Western blotting
Tissue samples were ground on ice, lysed thoroughly with RIPA lysis buffer (Sigma-Aldrich, R0278), and centrifuged at 4°C for 15 min at 12,000 × g to collect the supernatant. Protein concentration was determined using the BCA Protein Assay Kit (Thermo Scientific, 23225). Thirty micrograms of protein samples were subjected to 10% SDS-PAGE electrophoresis, with constant voltage electrophoresis at 80 V for 30 min, followed by constant voltage electrophoresis at 120 V until complete separation. Subsequently, proteins were transferred to PVDF membranes (Millipore, IPVH00010) at a constant voltage of 100 V for 1 h. After blocking the PVDF membranes with 5% skim milk for 1 h at room temperature, they were incubated overnight at 4°C with the following primary antibodies: anti-TRPV1 (Abcam, ab305299, 1:1000), anti-MCP-1 (Abcam, ab7202, 1:1000), anti-CD86 (Abcam, ab220188, 1:1000), anti-CD163 (Abcam, ab182422, 1:800), anti-CD206 (Abcam, ab64693, 1:600), anti-p38 MAPK (Cell Signaling Technology, #9212, 1:1000), anti-phospho-p38 MAPK (Cell Signaling Technology, #9211, 1:750), and anti-β-actin (Cell Signaling Technology, #4970, 1:1000). The next day, membranes were washed three times with TBST for 10 min each, followed by incubation with corresponding HRP-conjugated secondary antibodies (Cell Signaling Technology, anti-rabbit #7074 or anti-mouse #7076, 1:5000) at room temperature for 1 h, and washed again. Chemiluminescence was detected using ECL reagent (Yeasen, 36208ES60) and the membranes were imaged using a ChemiDoc Imaging System (Bio-Rad). Band densities were analyzed using Image J software (NIH) to calculate the relative expression levels of the target proteins normalized to β-actin.
Immunofluorescence staining
After dewaxing the DRG tissue sections, they were washed three times with PBS for 5 min each. Then, they were permeabilized with 0.25% Triton X-100 for 10 min and blocked with 5% BSA for 1 h. The corresponding primary antibodies: anti-F4/80 (Abcam, ab6640, 1:200 dilution), anti-CD86 (Abcam, ab53004, 1:100 dilution), and anti-CD206 (Abcam, ab64693, 1:150 dilution) were added and incubated at 4°C overnight. The next day, the sections were washed three times with PBS for 5 min each, followed by the addition of fluorescence-labeled secondary antibodies (Goat anti-Rabbit IgG Cross-Adsorbed Secondary Antibody, Alexa Fluor 488, Invitrogen, A-11008, 1:500 dilution; Goat anti-Rat IgG Cross-Adsorbed Secondary Antibody, Alexa Fluor 488, Invitrogen, A-11006, 1:500 dilution) and incubation at room temperature for 1 h. Nuclear staining was performed with DAPI (Yeasen, D 40728ES03, 1:10000 dilution) in the dark for 10 min, followed by another three washes with PBS. After sealing with an anti-fluorescence quenching agent (ProLong Gold Antifade Mountant, Invitrogen, P36930), the sections were observed under a fluorescence microscope (Leica DMi8, Leica Microsystems, Germany).
Flow cytometry
DRG tissues were collected, washed with pre-cooled PBS, minced, and digested in a digestion solution (0.25% trypsin + 0.1% collagenase) at 37°C for 30 min. The digested tissue was made into a single-cell suspension, filtered through a 400-mesh filter, and centrifuged at 1000 rpm for 5 min to collect the cells. The cells were resuspended in PBS, and Fc receptor blockers were added, followed by the addition of fluorescence-labeled antibodies such as F4/80, CD206, and CD86. The cells were incubated at 4°C in the dark for 30 min. After incubation, the cells were washed twice with PBS, centrifuged at 500 rpm for 5 min each time, and collected. The fluorescence signals of the cells were detected using a flow cytometer, and 10,000 cell data were collected. The proportions of F4/80+ macrophages, as well as M2 (CD206+) and M1 (CD86+) macrophages, were analyzed using FlowJo software.
Primary DRG extraction
Six- to eight-week-old male SD rats were used, and their DRG tissues were rapidly isolated and washed in pre-cooled PBS to remove surrounding fat and connective tissues. The DRG tissues were minced into approximately 1 mm3 pieces and enzymatically digested in a digestion solution containing 0.25% trypsin and 0.1% collagenase type IV at 37°C for 30 min with gentle agitation every 10 min. The digestion was terminated by adding DMEM (Gibco, 11965092) supplemented with 10% FBS (Gibco, 16000044). The digested tissue was triturated using a fire-polished glass Pasteur pipette to obtain a single-cell suspension, filtered through a 70 μm cell strainer (Corning, 352350), and then centrifuged at 200 × g for 5 min. The cell pellet was resuspended in DMEM containing 10% FBS, 1% penicillin-streptomycin (Gibco, 15140122), and 2 mM L-glutamine (Gibco, 25030081), and seeded onto poly-L-lysine-coated (Sigma-Aldrich, P4707, 0.1 mg/ml) culture dishes. Cells were maintained in a humidified incubator at 37°C with 5% CO2. After 24 h of culture, the non-adherent cells were removed by replacing the medium, and the adherent cells were cultured until reaching approximately 80% confluence (typically 3–4 days).
Cell culture
The murine macrophage cell line RAW264.7 (ATCC TIB-71) was retrieved from liquid nitrogen storage and rapidly thawed in a 37°C water bath until approximately 80% of the cell suspension was thawed. The cell suspension was then transferred to a 15 ml centrifuge tube containing 10 ml of pre-warmed DMEM high-glucose medium (Gibco, 11965092) and centrifuged at 200 × g for 5 min to remove the cryopreservation medium. The cell pellet was gently resuspended in complete culture medium consisting of DMEM high-glucose supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco, 16000044), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, 15140122). Cells were seeded in T75 culture flasks (Corning, 430641) at a density of 1 × 106 cells per flask and maintained in a humidified incubator at 37°C with 5% CO2. The culture medium was changed every 48 h, and cells were subcultured at 80%–90% confluence (approximately every 2–3 days) using cell scrapers (Corning, 3010) to detach the cells, as trypsin can alter macrophage surface receptors. For all experiments, cells between passages 5 and 15 were used to ensure consistency in cellular responses.
Macrophage-conditioned medium acquisition
RAW264.7 cells were cultured to 80% confluence in complete DMEM medium and divided into control and evodiamine-treated groups. The control group received an equal volume of vehicle (0.1% DMSO in culture medium), while the evodiamine-treated group was incubated with 10 μM evodiamine (Sigma-Aldrich, E3531, dissolved in DMSO with final DMSO concentration <0.1%) for 2 h at 37°C in a humidified 5% CO2 incubator. Subsequently, both groups were treated with 1 μM paclitaxel (Sigma-Aldrich, T7402) and incubated for an additional 24 h under standard culture conditions. Following incubation, the culture supernatants were carefully collected without disturbing the adherent cell layer using a sterile serological pipette. The collected media were centrifuged at 1000 rpm (approximately 200 × g) for 5 min at 4°C to remove cellular debris. The resulting supernatants, designated as macrophage-conditioned medium (MCM), were filtered through 0.22 μm sterile filters (Millipore, SLGP033RS) to ensure complete removal of cell debris and to maintain sterility. The MCM was then aliquoted into sterile cryovials (1 ml per vial) and stored at −80°C until further use. For experiments, frozen MCM aliquots were thawed on ice immediately before application to avoid repeated freeze-thaw cycles that could compromise bioactive factors.
Dual-luciferase reporter gene assay
First, reporter gene plasmids containing wild-type (WT) and mutant (MUT) TRPV1 promoter sequences were constructed (GeneChem, Shanghai). The TRPV1 promoter sequence was cloned into the promoter region of a dual-luciferase reporter vector (pGL3-luciferase reporter vector), placing the luciferase gene under the control of the TRPV1 promoter. The mutant (MUT) plasmid was generated by introducing mutations at critical binding sites within the TRPV1 promoter using site-directed mutagenesis. Subsequently, a constitutively phosphorylated p38 MAPK overexpression plasmid (OE-p-p38MAPK) was constructed by mutating threonine at position 180 and tyrosine at position 182 of the p38 MAPK protein to glutamic acid, with an empty vector plasmid (OE-NC) serving as control.
HEK 293T cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS) in a humidified incubator at 37°C with 5% CO₂. Culture medium was replaced regularly to maintain optimal cell growth conditions. HEK 293T cells were seeded at an appropriate density (5 × 10⁴ cells/well) in 24-well plates and cultured until reaching 70%–80% confluence. Following the transfection reagent protocol, TRPV1 promoter reporter plasmids (WT or MUT), OE-p-p38MAPK overexpression plasmids, and OE-NC plasmids were mixed with Lipofectamine™ 3000 transfection reagent (Thermo Fisher, L3000150), gently mixed, and incubated at room temperature for 20–30 min. The transfection mixture was added to cell culture wells, and plates were gently agitated to ensure uniform distribution. Fresh culture medium was replaced 4–6 h post-transfection to remove unbound transfection reagents.
Cells were lysed using cell lysis buffer (Promega, #E1941) 48 h post-transfection, and lysates were collected. Detection was performed using the Dual-Luciferase Reporter Assay System (Promega, #E1910). Firefly luciferase fluorescence intensity was measured first, followed by the addition of Stop & Glo reagent for Renilla luciferase detection. The ratio of firefly to Renilla luciferase activity was calculated to normalize for transfection efficiency differences, yielding relative TRPV1 promoter activity values.
Statistical analysis
All results are expressed as the mean ± standard deviation (SD). To compare quantitative data between experimental groups, independent sample t-tests, one-way analysis of variance (ANOVA) or two-way analysis of variance were used. p < 0.05 indicates a statistically significant difference between groups. Data analysis was performed using GraphPad Prism 9.5.0 software.
Results
Establishment of the paclitaxel-induced CIPN rat model
The paclitaxel-induced chemotherapy-induced peripheral neuropathy (CIPN) rat model was established by administering paclitaxel (2 mg/kg, intraperitoneal injection) according to the schedule outlined in Figure 1(a). Model establishment was assessed using standardized mechanical and thermal pain tests. The mechanical paw withdrawal threshold in response to von Frey filament stimulation exhibited a significant decrease beginning on day 3 post-paclitaxel administration, reaching its nadir on day 7, and remaining significantly depressed through day 21 (Figure 1(b)). Similarly, the paw withdrawal latency in response to noxious thermal stimuli demonstrated increased thermal hypersensitivity starting from day 3 after paclitaxel administration, with peak sensitivity observed on day 21 and sustained thereafter (Figure 1(c)). However, there was no significant difference in mechanical paw withdrawal threshold and paw withdrawal latency between the blank group and vehicle group. These behavioral alterations confirmed successful establishment of the paclitaxel-induced CIPN rat model. Furthermore, quantitative analysis of transient receptor potential vanilloid (TRPV) channel mRNA expression in dorsal root ganglia (DRG) revealed that on day 21 post-paclitaxel administration, Trpv1 and Trpv4 transcripts were significantly upregulated in the DRG of CIPN rats compared to controls (Figure 1(d)).

Establishment and characterization of the paclitaxel-induced chemotherapy-induced peripheral neuropathy (CIPN) rat model. (a) Schematic representation of the experimental timeline showing paclitaxel administration (2.0 mg/kg, i.p.), behavioral testing schedule, and tissue collection timepoints. (b) Development of mechanical allodynia as measured by paw withdrawal threshold (g) in blank (PBS), vehicle (DMSO) and paclitaxel-treated (PTX) rats over a 21-day period. (c) Development of thermal hyperalgesia as measured by paw licking latency (s) in blank, vehicle and PTX-treated rats. (d) Relative mRNA expression of TRPV channel subtypes (TRPV1, TRPV2, TRPV3, and TRPV4) in dorsal root ganglia (DRG) of blank, vehicle (DMSO) and PTX-treated rats at day 21.
Evodiamine alleviates paclitaxel-induced mechanical allodynia and thermal hyperalgesia in rats in a dose-dependent manner
To investigate the analgesic effects of evodiamine on paclitaxel (PTX)-induced CIPN, varying doses (0.3, 1, and 3 mg/kg) of evodiamine (Evo) were administered intraperitoneally to CIPN rats on day 21 post-PTX injection. Mechanical allodynia assessment demonstrated that evodiamine administration significantly attenuated PTX-induced mechanical hypersensitivity in a dose-dependent manner. The highest dose (3 mg/kg) of evodiamine increased the mechanical paw withdrawal threshold by more than 210% compared to the PTX + vehicle group (Figure 2(a)). The peak analgesic effect on mechanical sensitivity was observed 60 min post-evodiamine administration, persisted until 90 min, and dissipated by 120 min. In thermal hyperalgesia assessments, paw withdrawal latency in evodiamine-treated animals was significantly prolonged compared to the PTX + vehicle group, with approximately 190% increase in latency (Figure 2(b)). The analgesic effect of 3 mg/kg evodiamine reached maximal efficacy at 60 min and persisted for 120 min, although efficacy diminished rapidly after the 60-min peak. These observations indicate that evodiamine exerts a dose-dependent and time-limited analgesic effect on PTX-induced CIPN rats, ameliorating both mechanical and thermal hypersensitivity.
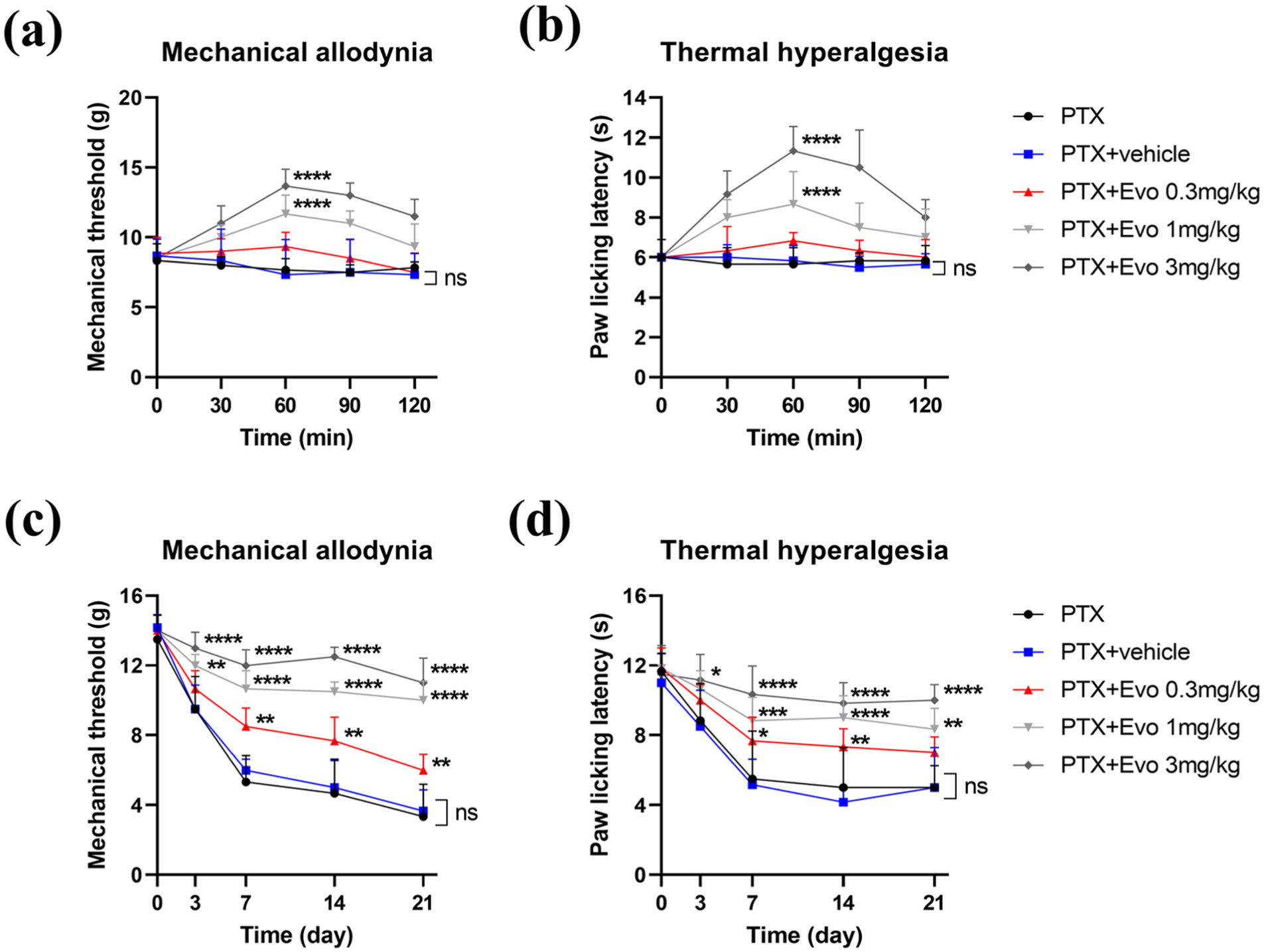
Evodiamine alleviates paclitaxel-induced mechanical allodynia and thermal hyperalgesia in rats in a dose-dependent manner. (a, b) Acute analgesic effects of evodiamine on mechanical allodynia (a) and thermal hyperalgesia (b) in in PTX, PTX + vehicle (DMSO) and PTX + evodiamine rats. On day 21 post-paclitaxel administration, rats received a single intraperitoneal injection of evodiamine at different doses (0.3, 1.0, or 3.0 mg/kg), and nociceptive responses were measured at 0, 30, 60, 90, and 120 min post-injection. (c, d) Preventive effects of evodiamine on the development of mechanical allodynia (c) and thermal hyperalgesia (d) in CIPN rats. Animals received daily intraperitoneal injections of evodiamine (1.0 or 3.0 mg/kg) or vehicle (DMSO) following paclitaxel (2.0 mg/kg, i.p.) administration, and pain behaviors were assessed over a 21-day period.
To further elucidate the preventive potential of evodiamine on the development of PTX-induced CIPN, rats received daily intraperitoneal administration of 0.3, 1, and 3 mg/kg evodiamine following PTX injection. Results demonstrated that mechanical allodynia was significantly attenuated in the PTX + evodiamine group compared to both PTX + vehicle and PTX groups (Figure 2(c)). Similarly, thermal paw withdrawal latency in the PTX + evodiamine group was significantly increased relative to both PTX + vehicle and PTX groups (Figure 2(d)). These findings suggest that evodiamine exhibits potent analgesic properties that can modulate both the development and progression of mechanical allodynia and thermal hyperalgesia in PTX-induced CIPN rats.
Evodiamine inhibits the upregulation of TRPV1 in the DRG of paclitaxel-induced CIPN rats, but has no effect on TRPV4
Given the established roles of TRPV1 and TRPV4 in nociception, we investigated whether evodiamine modulates pain through regulation of these channels by examining the mRNA expression of Trpv1 and Trpv4 in the DRG of paclitaxel-induced CIPN rats across experimental groups. Quantitative analysis revealed significant upregulation of both Trpv1 and Trpv4 transcripts in the DRG of paclitaxel-treated rats. Notably, evodiamine treatment selectively inhibited the upregulation of Trpv1 in paclitaxel-induced CIPN rats without affecting Trpv4 expression (Figure 3(a) and (b)). These data suggest that evodiamine may ameliorate CIPN specifically by inhibiting paclitaxel-induced TRPV1 upregulation in the DRG.

Evodiamine selectively inhibits the upregulation of TRPV1 but not TRPV4 in the DRG of paclitaxel-induced CIPN rats. (a) Relative mRNA expression levels of Trpv1 in dorsal root ganglia (DRG) tissues from rats treated with PTX, PTX + vehicle (DMSO) and PTX + evodiamine (0.3, 1.0, or 3.0 mg/kg, daily) for 21 days. Expression was normalized to Gapdh. (b) Relative mRNA expression levels of Trpv4 in DRG tissues from the same experimental groups, normalized to Gapdh.
Evodiamine protects CIPN rats in a TRPV1-dependent manner
To investigate whether TRPV1 participates in evodiamine’s protective effects against PTX-induced CIPN, rats were treated with the TRPV1 antagonist capsazepine and/or evodiamine (Evo). Experimental results demonstrated that in the PTX + Evo group, evodiamine treatment significantly reduced mechanical allodynia and thermal hyperalgesia in rats. In the capsazepine alone treatment group, both mechanical allodynia and thermal hyperalgesia were increased compared to the PTX + Evo group. Although the capsazepine + Evo combination treatment group showed some improvement in mechanical allodynia and thermal hyperalgesia, no significant differences were observed compared to the capsazepine alone intervention group (Figure 4(a) and (b)). These experimental results indicate that the analgesic effects of evodiamine may be primarily dependent on TRPV1.

Evodiamine alleviates paclitaxel-induced mechanical allodynia and thermal hyperalgesia in rats in a TRPV1-dependent manner. (a, b) Damage effects of TRPV1 on the development of mechanical allodynia (a) and thermal hyperalgesia (b) in CIPN rats. Animals received daily intraperitoneal injections of evodiamine (3.0 mg/kg) or CAZ following paclitaxel (2 mg/kg) administration, and pain behaviors were assessed over a 21-day period.
Evodiamine reduces macrophage infiltration and inflammatory responses in the DRG of paclitaxel-induced CIPN rats
Macrophage-mediated inflammation represents a critical component in CIPN pathogenesis. Flow cytometric analysis and immunohistochemical evaluation demonstrated that the population of F4/80-positive macrophages in the DRG was significantly reduced in evodiamine-treated animals compared to those receiving chemotherapy alone (Figure 5(a) and (b)). Furthermore, ELISA analysis revealed significant upregulation of pro-inflammatory cytokines TNF-α, IL-6, IL-1β, and MCP-1 in the DRG following paclitaxel administration (Figure 5(c)–(f)). Evodiamine treatment significantly attenuated this paclitaxel-induced upregulation of TNF-α, IL-1β, IL-6, and MCP-1 in DRG tissues (Figure 5(c)–(f)). These results suggest that evodiamine’s neuroprotective effects against paclitaxel-induced CIPN may be mediated through inhibition of macrophage infiltration and suppression of inflammatory cytokine expression in the DRG.

Evodiamine reduces macrophage infiltration in the DRG of paclitaxel-induced CIPN rats. (a) Flow cytometry analysis shows the distribution of CD11b and F4/80 double-positive macrophages in different treatment groups. n = 3 per rats group. (b) Immunofluorescence staining shows the expression of F4/80 (green) and DAPI (blue) in DRG tissue, with merged images showing their colocalization. n = 6 per rats group. (c–f) protein expression levels of TNF-α (c), IL-6(d), IL-1β(e), and MCP-1 (f) in rat DRG tissue is detected by ELISA. n = 6 per rats group.
Evodiamine enhances M2 polarization of macrophages in the DRG tissue of paclitaxel-induced CIPN rats
Disruption of the balance between pro-inflammatory M1 macrophages and anti-inflammatory/reparative M2 macrophages is implicated in neuroinflammatory pain conditions. We further investigated the mechanism by which evodiamine alleviates paclitaxel-induced CIPN by assessing M1 and M2 macrophage marker expression in DRG tissues across experimental groups. Immunofluorescence analysis revealed diminished CD86 (M1 marker) immunoreactivity in DRG tissues from evodiamine-treated animals compared to the chemotherapy group, with concurrent enhancement of CD206 (M2 marker) immunofluorescence signal (Figure 6(a) and (b)). These findings were corroborated by Western blot analysis, which demonstrated reduced expression of the M1 markers MCP1 and CD86 in DRG tissues from evodiamine-treated animals compared to the chemotherapy group, accompanied by increased expression of the M2 markers CD163 and CD206 (Figure 6(c)).

Evodiamine enhances M2 polarization of macrophages in the DRG tissue of paclitaxel-induced CIPN rats. (a) Immunofluorescence staining images show the expression of CD86 (green) in DRG tissue sections of different treatment groups, with DAPI (blue) used to mark nuclei. n = 6 per rats group. (b) Immunofluorescence staining images show the expression of CD206 (green) in DRG tissue sections of different treatment groups. n = 6 per rats group. (c) WB analysis shows the protein expression levels of MCP-1, CD86, CD163, and CD206 in DRG tissue of different treatment groups, with β-actin as the internal reference protein. Bar graphs quantitatively analyze the expression levels of MCP-1, CD86, CD163, and CD206 relative to β-actin. n = 3 per rats group.
Evodiamine reduces TRPV1 expression in DRG neurons by inhibiting paclitaxel-induced M1 polarization of macrophages
To elucidate the cellular mechanisms underlying evodiamine’s effects on DRG, we investigated whether evodiamine-mediated modulation of macrophage inflammation directly influences DRG neurons. RAW264.7 macrophages were treated with or without 10 μM evodiamine for 24 h following paclitaxel exposure, and macrophage-conditioned medium (MCM) was collected. Flow cytometric analysis confirmed that evodiamine treatment significantly increased the proportion of CD206-positive M2 macrophages while decreasing the population of CD86-positive M1 macrophages (Figure 7(a) and (b)). Subsequently, primary DRG neurons were cultured with the collected MCM. Quantitative PCR and Western blot analyses demonstrated that compared to control conditions, MCM derived from paclitaxel-treated RAW264.7 cells (MCM-PTX) significantly enhanced both mRNA and protein expression of TRPV1 in primary DRG neurons (Figure 7(c) and (d)). In contrast, treatment with MCM from evodiamine-treated macrophages (MCM-Evo) significantly reduced TRPV1 expression at both transcriptional and translational levels compared to the MCM-PTX group (Figure 7(c) and (d)). These findings indicate that evodiamine attenuates TRPV1 expression in DRG neurons by inhibiting paclitaxel-induced M1 macrophage polarization, thereby modulating chemotherapy-induced peripheral neuropathic sensitivity.
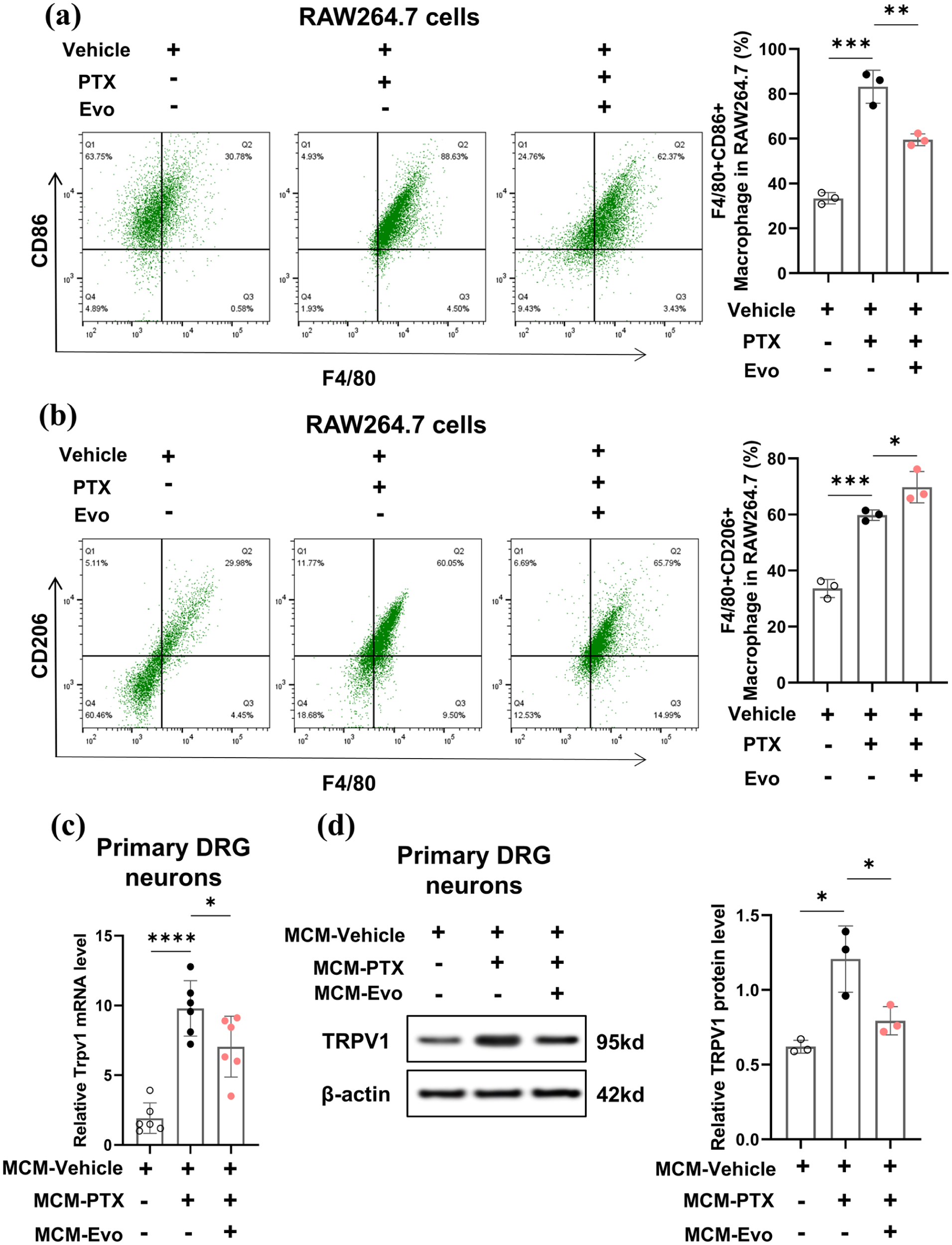
Evodiamine reduces TRPV1 expression in DRG neurons by inhibiting paclitaxelinduced M1 polarization of macrophages. (a) Flow cytometry analysis shows the percentage of macrophages marked by CD86 and F4/80 in RAW264.7 cells of different treatment groups. Quadrants Q2 (upper left quadrant) represent the F4/80+CD86+ macrophage population, respectively. n = 3 per group. (b) Flow cytometry analysis shows the percentage of macrophages marked by CD206 and F4/80 in RAW264.7 cells of different treatment groups. n = 3 per group. (c) Bar graphs show the relative expression levels of TRPV1 mRNA in different treatment groups. n = 6 per group. (d) WB analysis shows the protein expression levels of TRPV1 in different treatment groups. n = 3 per group.
Evodiamine mediates M2 polarization of macrophages to inhibit the p38 MAPK pathway in DRG neurons and downregulate TRPV1 expression
Previous research has established that activation of the p38 MAPK pathway can upregulate TRPV1 expression. 44 We investigated this mechanism by treating primary DRG neurons with macrophage-conditioned media. Western blot analysis revealed that exposure to MCM-PTX significantly elevated p38 MAPK phosphorylation in DRG neurons (Figure 8(a)–(c)). This increased phosphorylation was significantly attenuated in neurons treated with MCM-Evo compared to the MCM-PTX-treated group (Figure 8(a)–(c)). To confirm the role of p38 MAPK signaling in this process, we employed the selective p38 MAPK inhibitor SB203580, which effectively counteracted the MCM-PTX-induced activation of p38 MAPK signaling in DRG neurons (Figure 8(a)–(c)). These results indicate that evodiamine promotes M2 macrophage polarization, thereby inhibiting p38 MAPK pathway activation in DRG neurons and subsequently downregulating TRPV1 expression.

Evodiamine inhibits the p38 MAPK signaling pathway in DRG neurons to downregulate TRPV1 expression. (a) Representative Western blot images showing protein expression of TRPV1 (95 kDa), phosphorylated p38 MAPK (p-p38MAPK, 43 kDa), total p38 MAPK (40 kDa), and β-actin (42 kDa) in primary DRG neurons treated with different conditioned media. n = 3 per group. (b) Quantitative analysis of TRPV1 protein expression relative to β-actin. n = 3 per group. (c) Quantitative analysis of the phosphorylated p38 MAPK to total p38 MAPK ratio. Primary DRG neurons were treated with control medium (DMSO), macrophage-conditioned medium from paclitaxeltreated cells (MCM-PTX), macrophage-conditioned medium from evodiamine-treated cells (MCMEvo), or MCM-PTX plus the selective p38 MAPK inhibitor SB203580 (10 μM). n = 3 per group. (d) The constructed plasmid of sustained phosphorylation overexpression of p38MAPK (OE-p-p38MAPK) and the empty vector plasmid (OE-NC) were transfected into HEK 293T cells. The protein expression levels of p-p38MAPK in HEK 293T cells between the OE-NC group and OE-p-p38MAPK group were detected by WB. (e) The effect of p-p38MAPK overexpression on TRPV1 transcription in HEK 293T cells was detected by dual-luciferase reporter gene assay. n = 6 per group.
After confirming that p38 MAPK promotes TRPV1 expression, we further investigated whether p38 MAPK regulates TRPV1 expression at the transcriptional level. We constructed constitutively phosphorylated p38 MAPK overexpression plasmids (OE-p-p38MAPK) and empty vector plasmids (OE-NC) and transfected them into HEK 293T cells. Western blot results showed that compared to the OE-NC group, p-p38MAPK protein expression levels were significantly upregulated in HEK 293T cells transfected with OE-p-p38MAPK plasmids, indicating successful construction of the constitutively phosphorylated p38 MAPK plasmid (Figure 8(d)). Next, we used Lipofectamine 3000 to transiently transfect HEK 293T cells with wild-type pGL3-TRPV1 promoter (WT) constructs, mutant pGL3-TRPV1 promoter (MUT) constructs, and either OE-NC or OE-p-p38MAPK plasmids. After 48 h, firefly and Renilla luciferase activities were detected using the dual-luciferase reporter assay system. Dual-luciferase reporter gene assay results indicated that compared to the OE-NC group, relative luciferase activity was significantly increased in HEK 293T cells transfected with OE-p-p38MAPK plasmids, indicating upregulated TRPV1 promoter transcriptional activity. However, in HEK 293T cells transfected with mutant TRPV1 promoter constructs, luciferase activity in cells transfected with OE-p-p38MAPK plasmids showed no significant difference compared to the OE-NC group (Figure 8(e)). These data suggest that p-p38MAPK can upregulate TRPV1 expression at the transcriptional level.
Depletion of macrophages in CIPN rats with clodronate liposomes inhibits activation of the p38 MAPK pathway and upregulation of TRPV1 in DRG tissue
Protein expression analysis of DRG tissues revealed striking differences between treatment groups. Compared to paclitaxel-treated rats, animals receiving combined paclitaxel + evodiamine treatment exhibited significantly reduced TRPV1 expression, accompanied by markedly decreased phosphorylation of p38 MAPK (Figure 9). To confirm the critical role of macrophages in this pathway, we employed clodronate liposomes for selective macrophage depletion. Notably, rats treated with the combination of paclitaxel + clodronate liposomes displayed significantly decreased TRPV1 expression in DRG tissues, coupled with reduced activation of the p38 MAPK signaling pathway compared to rats receiving only paclitaxel + evodiamine (Figure 9). These compelling results establish a mechanistic link whereby chemotherapy-induced M1 macrophage polarization drives p38 MAPK phosphorylation in DRG tissues, subsequently promoting TRPV1 upregulation in DRG neurons.
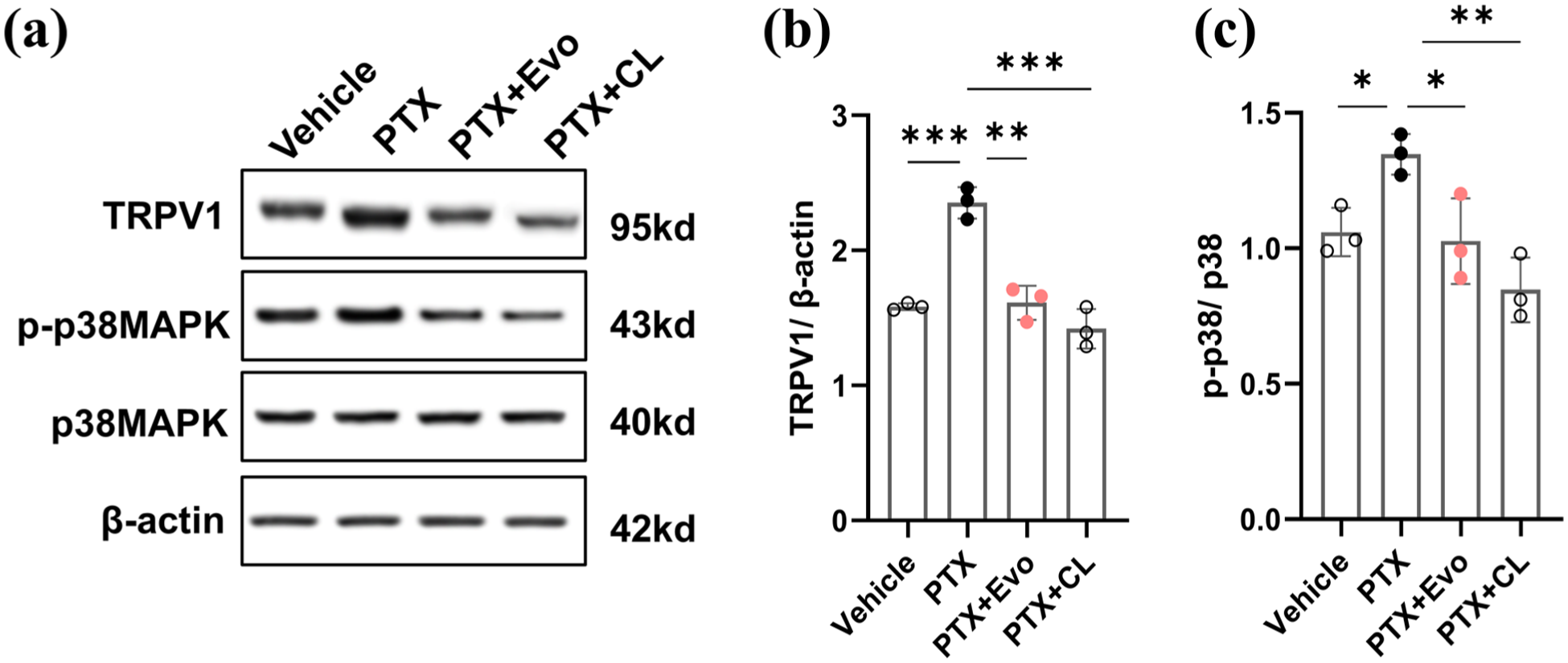
Depletion of macrophages with clodronate liposomes inhibition of the p38 MAPK pathway and TRPV1 expression in DRG tissue. (a) Representative Western blot images showing protein expression of TRPV1, phosphorylated p38 MAPK (p-p38MAPK), total p38 MAPK, and β-actin in DRG tissues from different treatment groups. (b) Quantitative analysis of TRPV1 protein expression relative to β-actin. (c) Quantitative analysis of the phosphorylated p38 MAPK to total p38 MAPK ratio. Treatment groups include vehicle (DMSO), paclitaxel alone (PTX), paclitaxel plus evodiamine (PTX+Evo), and paclitaxel plus evodiamine and clodronate liposomes (PTX+CL).
Discussion
Chemotherapy-induced neuropathic pain (CINP) represents a common, severe, and persistent complication of cancer treatment characterized by peripheral neuropathy. 45 With increasing cancer incidence, improved survival rates, and enhanced cure rates, CINP prevalence continues to rise. 46 Our findings demonstrate that evodiamine significantly attenuates CINP symptomatology by inhibiting pro-inflammatory cytokine release and TRPV1 upregulation in the dorsal root ganglia (DRG) of paclitaxel-treated rats. Mechanistically, evodiamine promotes macrophage polarization toward an M2 phenotype and suppresses p38/MAPK signaling pathway activation in DRG neurons, thereby downregulating TRPV1 expression. These results suggest that evodiamine may ameliorate CINP by modulating the balance between M1 and M2 macrophage polarization in neuropathic pain states.
TRPV1 and TRPV4 function as sensory receptors for noxious stimuli,31,47 and previous studies have demonstrated that paclitaxel administration induces upregulation of both channels.32,48 Evodiamine has been shown to mitigate bortezomib-induced peripheral neuropathy by inhibiting MAPK signaling, thereby reducing oxidative stress and ferroptosis. 49 Previous research also indicates that evodiamine alleviates paclitaxel-induced neuropathic pain by attenuating inflammation and preserving mitochondrial antioxidant function. 17 In the present study, we observed upregulation of both Trpv1 and Trpv4 mRNA in affected rat DRG. Notably, evodiamine selectively inhibited Trpv1 upregulation without affecting Trpv4 expression. These findings suggest that evodiamine’s analgesic effects in paclitaxel-induced CINP may be specifically mediated through TRPV1 channel modulation.
The TRPV1 channel is widely distributed in the somatosensory system, particularly in primary afferent neurons, where it modulates nociceptive signaling from the DRG and spinal cord. The p38/MAPK-TRPV1 signaling axis plays a pivotal role in pain signal transduction and inflammatory responses.50–53 Our results demonstrate that paclitaxel treatment significantly enhances p38 MAPK phosphorylation in DRG neurons while concurrently upregulating TRPV1 expression. Evodiamine administration significantly reduces p38 MAPK phosphorylation and inhibits TRPV1 upregulation. Pharmacological blockade of p38 MAPK signaling with SB203580 counteracts the pathway activation induced by evodiamine. Numerous experimental investigations have established that enhanced TRPV1 channel expression in DRG neurons contributes to development of various sterile inflammatory pain models across different pathological conditions.27–29 Collectively, these results indicate that evodiamine exerts analgesic effects by inhibiting p38 MAPK signaling pathway activation and downregulating TRPV1 expression. P38 MAPK can regulate protein expression by activating transcription factors such as cAMP response element-binding protein (CREB), ETS homolog transcription factor (ELK-1), and eukaryotic translation initiation factor 4E (eIF4E), thereby enhancing downstream target gene mRNA transcriptional activity, stability, or translation processes.54–57 We explored the regulatory relationship between p38 MAPK and TRPV1 through dual-luciferase reporter experiments. Our dual-luciferase reporter experimental results suggest that p38 MAPK can upregulate TRPV1 protein expression by enhancing TRPV1 transcriptional levels. Previous research evidence also indicates that p38 MAPK is essential for TRPV1 protein upregulation, 44 and our evidence demonstrates that evodiamine increases TRPV1 protein expression in DRG in a manner dependent on p38 MAPK activation.
It is worth noting that in addition to p38 MAPK, pro-inflammatory cytokines (such as TNF-α, IL-1β, and IL-6) can also activate ERK and JNK signaling pathways, which play important roles in DRG neuronal excitability and pain sensitization. Activation of ERK and JNK leads to multiple downstream effects, including upregulation and sensitization of TRPV1 channels, thereby exacerbating pain responses. In this study, we primarily focused on evodiamine’s regulatory effects on the p38 MAPK-TRPV1 axis. Although we did not specifically examine ERK and JNK phosphorylation levels, previous research results suggest that evodiamine may have broad regulatory effects on the entire MAPK family activity.14,49 Evodiamine’s anti-inflammatory properties on macrophages may not be limited to p38 MAPK inhibition but may also exert synergistic anti-inflammatory effects through simultaneous regulation of other MAPK family members. Therefore, we speculate that evodiamine may achieve comprehensive pain modulation through multiple collaborative mechanisms. We acknowledge that although this study did not directly examine ERK and JNK, through existing data and literature support, we can propose evidence-based hypotheses that evodiamine may also have regulatory effects on these pathways. We will further validate this hypothesis in future research to more comprehensively elucidate its analgesic mechanisms.
Elevated TNF-α and IL-6 levels are well-established contributors to pain states, as their upregulation in key neuroanatomical regions including the spinal cord, anterior cingulate cortex, and DRG promotes pain development.58–60 Evodiamine possesses anti-inflammatory properties relevant to cancer treatment. 61 Our study revealed that evodiamine significantly reduces F4/80+ macrophage infiltration in the DRG of paclitaxel-induced CINP rats, decreases expression of M1 macrophage markers MCP1 and CD86, and increases expression of M2 macrophage markers CD163 and CD206. These findings suggest that evodiamine attenuates DRG inflammation by promoting macrophage polarization toward an M2 phenotype, thereby alleviating CINP symptoms. Additionally, evodiamine significantly reduces TNF-α, IL-1β, IL-6, and MCP-1 expression in the DRG, further substantiating its anti-inflammatory activity. These data indicate that evodiamine alleviates mechanical allodynia and thermal hyperalgesia in CINP rats by facilitating macrophage phenotypic transition from pro-inflammatory to anti-inflammatory states. By modulating TNF-α and IL-6 expression in the DRG, evodiamine may regulate neuroinflammatory responses associated with neuropathic pain. This mechanism underscores evodiamine’s potential as a therapeutic agent for managing CINP-associated neuropathic pain.
Research indicates that evodiamine inhibits neuroinflammation through the AKT/Nrf2/HO-1/NF-κB axis, 16 and this mechanism may have connections with the novel mechanism of alleviating CIPN through M2 macrophage polarization and the p38/MAPK-TRPV1 axis. On one hand, activation of the AKT/Nrf2/HO-1 axis inhibits NF-κB and suppresses the release of pro-inflammatory factors (such as TNF-α, IL-6, IL-1β), which is highly consistent with the anti-inflammatory properties of the M2 phenotype during macrophage polarization. NF-κB is a key regulator of inflammatory responses, and its activation leads to increased expression of TNF-α, IL-6, and IL-1β while promoting macrophage polarization toward the M1 phenotype. By inhibiting the NF-κB pathway, evodiamine can effectively reduce neuroinflammation. More importantly, evodiamine’s ability to block the TLR4-NF-κB signaling pathway has been demonstrated. 51 These studies all indicate that evodiamine may exert its anti-inflammatory effects through multiple targets. Furthermore, the classical TLR4-NF-κB and TLR4-MAPK pathways are crucial in macrophage-mediated inflammation initiation. From this, we can reasonably speculate that the AKT/Nrf2/HO-1/NF-κB axis may also inhibit the p38/MAPK-TRPV1 axis through macrophage-mediated actions, directly reducing neuronal excitability and thereby indirectly alleviating neuroinflammatory pain. These two mechanisms may work synergistically or as cascading reactions to collectively alleviate CIPN symptoms. Future research directions could further explore the molecular connections between these two mechanisms, as well as their differential effects and clinical application value.
Additionally, evodiamine’s activation of the AKT/Nrf2/HO-1 axis and subsequent NF-κB inhibition can promote microglial polarization toward the M2 phenotype. 16 This may be related to evodiamine’s protective effects on mitochondrial function and its antioxidant properties. 17 As a transcription factor, Nrf2 can activate the expression of a series of antioxidant genes, including HO-1. HO-1 not only possesses antioxidant functions but also influences inflammatory responses by regulating macrophage function. Furthermore, mitochondria serve not only as cellular energy factories but also play important roles in regulating intracellular reactive oxygen species (ROS) levels. In CIPN models, mitochondrial dysfunction leads to excessive ROS generation, subsequently triggering oxidative stress and inflammatory responses. Increased ROS can activate multiple signaling pathways, among which the p38 MAPK pathway plays a central role in pain sensitization. Activation of p38 MAPK leads to a cascade of downstream signaling reactions, ultimately resulting in TRPV1 channel upregulation and sensitization. Therefore, we speculate that evodiamine may also regulate mitochondrial function through AKT/Nrf2/HO-1 to reduce ROS generation, thereby inhibiting p38 MAPK activation and ultimately indirectly regulating TRPV1 expression. Although we did not conduct specific experiments to verify whether evodiamine regulates TRPV1 through this particular cascade reaction, previous studies and existing data support this possibility. This mechanism provides a new perspective for understanding evodiamine’s analgesic effects and offers potential directions for future research.
It is important to acknowledge the limitations of this investigation to contextualize its findings appropriately. Our study of evodiamine’s effects on paclitaxel-induced CINP did not evaluate certain pathophysiological aspects including peripheral nerve terminal degeneration, neural structural alterations, and demyelination. These limitations present opportunities for future investigations to more comprehensively characterize CINP mechanisms. Examination of peripheral nerve terminal loss, neural tissue structural changes, and demyelination processes would provide valuable insights into CINP pathophysiology and potentially elucidate additional mechanisms through which evodiamine exerts its therapeutic effects.
Conclusion
Our findings demonstrate that evodiamine effectively alleviates chemotherapy-induced peripheral neuropathy symptoms through multifaceted mechanisms. Specifically, evodiamine inhibits TRPV1 channel upregulation and attenuates pro-inflammatory cytokine release in the dorsal root ganglia of paclitaxel-treated rats. Mechanistically, evodiamine promotes macrophage polarization toward an anti-inflammatory M2 phenotype while suppressing the p38/MAPK signaling pathway in DRG neurons, resulting in decreased TRPV1 expression and reduced nociceptive signaling. The selective modulation of TRPV1 channels, without affecting TRPV4, suggests a targeted mechanism underlying evodiamine’s analgesic properties. These results not only elucidate the molecular pathways through which evodiamine mitigates chemotherapy-induced neuropathic pain but also establish a foundation for developing novel therapeutic strategies targeting macrophage polarization and TRPV1 signaling in CIPN management. Further investigations into evodiamine’s potential clinical applications may offer promising alternatives for patients experiencing this debilitating side effect of cancer treatment.
Footnotes
Author contributions
The contributions of the authors are as follows: Peipei Wu, Yong Chen, Kequn Xu, Qiangqiang Zhou, Zhourui Li, and Rong Yang collected the data for this study. Peipei Wu, Yong Chen, Kequn Xu, and Qiang Jiang analyzed and interpreted the data. Qiangqiang Zhou, Zhourui Li, and Rong Yang critically revised the manuscript and provided significant intellectual input. All authors have approved the final version of the manuscript for publication.
Data availability statement
The data supporting the results of this study can be obtained from the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Science and Technology Project of Changzhou Health Commission (No. QN202231).
Ethical considerations
All animal experiments conducted in this study were approved by the Animal Welfare Committee of the Second People’s Hospital of Changzhou Affiliated to Nanjing Medical University.