
Abstract
Neuropathic pain (NP), a specific subtype of chronic pain, can induce depression-like behavior, presenting significant challenges for clinical treatment. Ascorbic acid (AA) is a free radical scavenger; however, its regulatory effects on NP, particularly within the spinal cord, remain ambiguous. In this research, we examined the impact of AA on NP and associated depression-like behavior by establishing a spinal nerve injury (SNI) NP model. Behavioral tests showed that mice in the SNI group exhibited hyperalgesia and depression-like behavior. Compared with the control group, the SNI group showed attenuated antioxidant responses (impaired Nrf2 signaling), excessive NLRP3 inflammasome activation, and elevated AMPK activity in spinal cord tissues. However, treatment with AA alleviated NP and depression-like behavior in mice with SNI by suppressing NLRP3-mediated inflammation and enhancing Nrf2-driven antioxidant responses. In vivo electrophysiology demonstrated that AA reversed the increase in theta, alpha, and beta band energies in mice with SNI. The results suggest that AA mitigates NP and comorbid depression-like behavior by inhibiting the activity of NLRP3 inflammasome and activating the Nrf2 pathway. Its ability to normalize neurophysiological rhythms further supports its therapeutic potential for NP. These findings imply that AA is a novel therapeutic agent for NP.
Introduction
Neuropathic pain (NP) results from damage or dysfunction within the somatosensory system. 1 It can be triggered by both harmful and non-toxic stimuli, leading to an exaggerated pain response that manifests as spontaneous, persistent, or radiating sensations. 2 Chronic NP can exacerbate symptoms, resulting in irritability and restlessness and potentially contributing to psychological conditions such as anxiety and depression. 3 Given the complexity of NP, the primary objectives of existing treatment strategies are to manage pain and address comorbidities, such as depression. 4 These strategies often involve the combined use of antidepressants and nonsteroidal anti-inflammatory drugs. Although traditional treatments improve patients’ quality of living to some extent, it is noteworthy that more than two-thirds of patients with NP fail to obtain adequate relief. 5 Consequently, there is a pressing necessity for research into more potent treatment modalities to alleviate NP and symptoms of anxiety and depression.
Harmful signals are transmitted through primary afferent fibers (Ab) to the spinal cord and the brain, resulting in pain. 6 The spinal cord is crucial for the incorporation of pain information and the phenomenon of central neural pain sensitization. 7 NP induces both pathological and physiological alterations in the spinal cord. It is capable of initiating the NLRP3 inflammasome, which upregulates the activity of caspase-1 and the pro-inflammatory cytokine IL-1β, 8 together with lead to an increase in oxidative stress responses. 9 Oxidative stress within the spinal cord significantly contributes to neuroinflammation and NP. 10 In animal models of NP, increased oxidative stress has been proved to set off multiple transcription factors while decreasing Nrf2 expression. 11 The activation of transcription factors results in unequal expression of genes, provoking inflammatory responses 12 and leading to anxiety- and depression-like behaviors over time. 13 Ascorbic acid (AA) has been shown to stimulate the signaling pathway of AMP-activated protein kinase (AMPK), which lessens the activity of dorsal root ganglion neurons and alleviates NP. 14 Consequently, this signaling pathway serves as a promising therapeutic target for NP. 15 Hence, further research is indispensable to clarify the specific pathological mechanisms responsible for NP.
AA, a natural antioxidant, can effectively eliminate various reactive oxygen species, whereupon safeguarding cells against oxidative damage. 16 Owing to its anti-inflammatory properties, it can decrease the concentrations of pro-inflammatory cytokines such as tumor necrosis factor-alpha, interferons, and interleukins. 9 Additionally, AA has demonstrated potential antidepressant effects.17,18 In this research, we successfully established a mouse model of sciatic nerve injury (SNI) and conducted analyses focusing on mouse behavior, tissue morphology, and protein expression levels to examine the role and mechanisms of AA in NP and associated depression-like behavior. The findings provide theoretical and empirical support for the use of AA in the treatment of NP and depression.
Materials and methods
Animals
Male C57BL/6 mice, aged between 6 and 8 weeks with a body weight of 20 ± 2 g, were sourced from the Hubei Experimental Animal Center. These mice were kept in a 12-h light and 12-h dark cycle, with continuous access to both food and water. All experimental procedures adhered to local and international ethical guidelines regarding animal usage, and significant measures were taken to reduce both the quantity of animals involved and their suffering.
Reagents and antibodies
Total and cleaved caspase-1 mouse antibody (catalog number: M025280) was obtained from Abmart. Antibody against Caspase-1 from rabbit (catalog number: AF5418), antibody against IL-1β from mouse (catalog number: BF8021), rabbit-derived antibody specific for NLRP3 (catalog number: DF15549), rabbit-derived β-actin antibody (catalog number: AF7018), rabbit-derived antibody specific for AMPK (catalog number: AF6423), and antibody from rabbit specific to P-AMPK (catalog number: AF3423) were acquired from Affinity Biosciences. Antibody against Nrf2 from rabbit (catalog number: A1244) was acquired from ABclonal Technology. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (H+L; AS014), the secondary antibody for western blotting, was procured from ABclonal Technology. For immunofluorescence analysis, the secondary antibody was obtained from Abcam. The hematoxylin–eosin (HE) staining solution (catalog number: BL700A) was gotten from Biosharp Life Sciences, and AA was acquired from Sigma (catalog number: 1043003).
Establishment of a mouse model of SNI and drug administration
After a period of 5 days dedicated to acclimatization, the mice involved in the study were randomly assigned into the control (CON), SNI, and SNI + AA groups, with seven mice allocated to each group. To establish the SNI model, the mice underwent anesthesia using isoflurane. The skin at the surgical site was disinfected with 75% alcohol, and an electric shaver was used to scrape the fur slightly from the knee down to the buttocks. A slit was made to reveal the sciatic, sural, tibial, and common peroneal nerves. The tibial and common peroneal nerves were tied using a 6.0 silk thread, while the sural nerve should be spared. Finally, the skin was sutured. Pain behavioral evaluations were carried out prior to the surgical procedure on day 0 and were repeated on days 7, 14, 21, 22, 23, 24, 25, 26, and 27 after the surgery.
From day 8 after establishing the SNI model, mice in the SNI + AA group underwent intraperitoneal injections of AA (10 mg/kg) for 21 days. Considering that AA has extremely poor chemical stability and is prone to degradation due to oxidation,19,20 we dissolved AA in DMSO and then diluted it to 2 mg/mL with 0.9% NaCl to maintain its stability.21,22 Four hours after the AA had been administered, the behavioral evaluations were performed. On day 28, all mice were sacrificed for subsequent experiments.
Behavioral tests
Mechanical threshold test
In order to facilitate acclimatization to their new surroundings, the mice were housed in a transparent glass enclosure for a duration of 30 min. Following this adaptation phase, Von Frey filaments were employed to exert pressure directed perpendicularly onto the plantar surface of the left hind paw. The filaments were applied for 2–3 s until a bent was observed. Drawing in of the hind paw and foot licking were marked as positive reactions. If a positive reaction occurred, the adjacent lower-level von Frey filaments were stimulated again. If a negative reaction occurred, the adjacent higher-level filaments were stimulated again. The positive and negative withdrawal reaction patterns were translated to 50% pain withdrawal threshold (PWT).
Hot plate test
The mice were carefully positioned onto a hot plate, which was maintained at 55 ± 1°C. The latency period until the mice exhibited behaviors such as foot-lifting, paw-licking, or jumping away from the heat source was noted. After a reaction was observed, the mice were immediately removed from the hot plate. The retention time of mice stayed on the hot plate was limited to 25 s to prevent tissue damage.
Tail suspension test (TST)
The tail of each mouse was attached to a purpose-made needle tube and suspended from a bracket 20 to 25 cm above the table surface. A white background was used to provide optimal contrast for imaging. The test duration was 6 min, with the first 2 min typically regarded as an adaptation period during which no data were recorded. The time spent being immobile and the time spent struggling to escape during the subsequent 4 min were recorded.
Forced swim test (FST)
A cylindrical water container was cleaned thoroughly to get rid of animal odors. The container, which had a total volume capacity of 5000 mL, was filled with 15 cm of water at a temperature of 25°C. Each mouse was introduced into the container, where its swimming behavior was monitored for a duration of 6 min. The times spent immobile and swimming by the mouse during the final 4 min were noted. After 6 min, the mouse was removed from the water and dried.
Sucrose preference test (SPT)
The SPT contains two stages: adaptation and experimentation. During the adaptation stage, two bottles with 1% sucrose solution and water were placed into each home cage. The position of the bottle was changed every 12 h. This stage lasted 2 days; after which, the mice were starved and dehydrated for 24 h. During the subsequent experimentation stage, the mice were allowed unhindered access to the sucrose solution and water for 2 h. Changes in the quantity of the sucrose solution and regular water were documented. The sucrose preference index of each mouse was derived from the following formula: sucrose consumption/(sucrose consumption + water consumption) * 100%.
HE staining
The mice were rendered unconscious using 10% chloral hydrate and subsequently perfused initially with 0.9% NaCl via the heart, followed by 4% paraformaldehyde until they became stiff. After perfusion, the spinal cord was removed and subjected to fixation. It was then embedded and sectioned into slices that measured 4 µm in thickness, using a microtome RM 2165 (manufactured by Leica Microsystems GmbH). The slices underwent a staining process according to the standard HE staining protocol and observed (Olympus IX73; Olympus). The ImageJ 1.51 j8 software (National Institutes of Health) was utilized to evaluate the images.
Immunofluorescence analysis
Intact spinal cord tissue sections that had been previously dewaxed were handled with 3% hydrogen peroxide. Afterwards, the sections were exposed to primary antibodies targeting IL-1β, Nrf2, NLRP3, caspase-1 and incubated overnight. Subsequently, secondary antibodies were introduced for a 1-h incubation and examined.
Western blotting
The mice were euthanized with 10% chloral hydrate. Tissues from the lumbar spinal cord were harvested, then kept on ice for homogenization. The tissues underwent centrifugation, then the supernatant was gathered and combined with 1/3 volume of 4× loading buffer. Total proteins extracted from each sample were quantified using a BCA protein assay kit. Proteins were fractionated by means of sodium dodecyl sulfate–polyacrylamide gel electrophoresis, after which the proteins were transported to a PVDF membrane. Then, the membrane was treated with 10% skim milk powder before being incubated overnight with primary antibodies against IL-1β (1:1000), Nrf2 (1:1000), AMPK (1:1000), p-AMPK (1:1000), NLRP3 (1:1000), and cleaved-caspase-1 (1:1000). On the following day, the membrane was incubated with a suitable secondary antibody for 1 h. The grayscale values were examined using ImageJ 1.51j8 software, with β-actin serving as a loading control.
Molecular docking
The AMPK protein structure (ID 4CFH) was obtained from PDB (https://www.rcsb.org/), whereas the three-dimensional structure of AA was derived from PubChem (https://pubchem.ncbi.nlm.nih.gov/). The AutoDock Vina (version 1.2.0) software (Center for Computational Structural Biology) was applied for molecular docking of AMPK and AA. The results were visualized using PyMOL. 23
Measurement of superoxide dismutase activity
The activity of superoxide dismutase (SOD) was measured using Cu/Zn-SOD and Mn-SOD assay kits with WST-8 (Beyotime, Shanghai, China). Mouse lumbar spinal cord tissues were first homogenized and centrifuged. The resulting supernatant was carefully collected and subsequently combined. The mixture was incubated to facilitate the reaction. Finally, the absorbance of each individual sample was quantified. SOD activity was quantified as units of total protein per milligram (U/mg protein).
Electrophysiological examination
Electrophysiological examination was carried out in accordance with the protocols described in previous studies.24,25 The mice received anesthesia from isoflurane and firmly positioned on a stereotactic device. A microdrive, specially designed to hold eight adjustable tetrodes, was implanted within the spinal cord. Before the implantation, the tips of the wires were electroplated using gold by applying a cathodal current, whereas the tetrodes were immersed in a gold solution, resulting in the reduction of impedance to 300–400 kΩ at a frequency of 1 kHz. A midline incision was carefully made along the left lumbar vertebrae until the spinal cord became visible through the intervertebral spaces located between the T12 and T13 vertebrae. Subsequently, tetrodes were inserted into the L3–L4 vertebrae to a depth of 250 mm beneath the dura utilizing the stereotactic frame while exercising caution to avoid the posterior spinal arteries. Wires were connected to a 32-channel connector (Omnetics Connector). Local field potentials (LFPs) were obtained through the recording of single electrode of each tetrode with a sampling frequency of 1 kHz. The acquired signals were filtered between 1 Hz and 400 Hz, notch filtered (notch width set to 0.1 Hz) to reduce 50-Hz AC noise, and subsequently stored on a computer for further offline evaluation. A multi-taper approach (five tapers) was used to construct LFP spectrograms.
Statistical analysis
Statistical evaluation was conducted via SPSS software (version 26.0). Data for the behavioral assessments were presented as mean ± standard error of the mean (SEM). The remaining data were expressed as the mean ± standard deviation (SD). Differences between groups with p-value less than 0.05 were deemed statistically significant.
Results
AA relieved pain in mice with SNI
SNI was induced to establish a mouse model of NP (Figure 1(a)). To evaluate pain caused by SNI, we used the Hargreaves and von Frey tests to detect thermal and mechanical pain thresholds, respectively. The consequences showed that the latency period of thermal pain was markedly lower in the SNI group (6.05 ± 0.22) than in the CON group (12.11 ± 0.33; p < 0.05; Figure 1(c)). Similarly, the mechanical pain threshold was notably lower in the SNI group (0.21 ± 0.05) than in the CON group (0.75 ± 0.18; p < 0.05; Figure 1(d)). These results indicated the mouse model of SNI-induced NP has been successfully established. To assess the analgesic properties of AA in the context of NP, mice suffering from SNI received an intraperitoneal injection of AA at a dosage of 10 mg/kg for 21 days from day 8 of model construction (Figure 1(b)). Treatment with AA significantly increased the latency period of thermal pain (SNI: 5.54 ± 0.11; SNI + AA: 11.02 ± 0.10; p < 0.05; Figure 1(c)–(c-4)) and the mechanical pain threshold (SNI: 0.21 ± 0.07; SNI + AA: 0.38 ± 0.07; p < 0.05) in mice with SNI (Figure 1(d)–(d-4)). These results indicate that intraperitoneal injection of AA relieves thermal pain and mechanical hyperalgesia caused by NP.

Effects of AA on neuropathic pain in mice with SNI. (a) Establishment of a mouse model of SNI. (b) Schematic diagram of the experimental procedures. After 5 days of acclimatization, the mice were used to establish an NP model (day 0). Behavioral tests for assessing pain threshold were performed on days 0, 7, 14, and 21. Behavioral tests for assessing depression-like behavior were performed on day 28. AA was intraperitoneally injected for 21 days beginning from day 8, and behavioral tests were conducted 4 h after AA administration. The mice were euthanized, and spinal cord tissues were collected for further analysis. (c, d) Changes in PWL (c) and PWT (d) values in the CON, SNI, and SNI + AA groups. Histograms demonstrating the PWL (c1–c4) and PWT (d1–d4) values on days 0, 7, 14, and 21 are shown. All data are presented as the mean ± SEM (n = 7). *p < 0.05 compared with the control group; #p < 0.05 compared with the SNI group.
AA alleviated depression-like behavior in mice with SNI
Chronic NP is often closely associated with depression-like behavior. 26 The SPT, along with the FST and TST, are widely recognized methodologies employed in the assessment of depression-like behaviors in animal subjects. 27 We used these tests to investigate the influences of AA on depression-like behavior in mice with SNI. SPT showed that sucrose consumption (CON: 1.85 ± 0.14; SNI: 1.14 ± 0.14; p < 0.05) and sucrose preference (CON: 0.57 ± 0.03; SNI: 0.36 ± 0.02; p < 0.05) were drastically lower in the SNI group than in the CON group (Figure 2(a), (b)). Furthermore, the immobility time of mice increased notably in the SNI group compared with that in the CON group in both TST (CON: 107.85 ± 11.15; SNI: 161.42 ± 9.88; p < 0.05; Figure 2(c), (d)) and FST (CON: 95.42 ± 19.86; SNI: 159 ± 10.49; Figure 2(e), (f)). However, treatment with AA significantly increased sucrose consumption (SNI: 1.14 ± 0.14; SNI + AA: 2.14 ± 0.14; p < 0.05) and sucrose preference (SNI: 0.36 ± 0.02; SNI + AA: 0.55 ± 0.02; p < 0.05) and significantly decreased the immobility time in both TST (SNI: 159 ± 10.49, SNI + AA: 66 ± 10.59; p < 0.05) and FST (SNI: 161.42 ± 9.88; SNI + AA: 109.85 ± 6.78) in mice with SNI (Figure 2). These findings indicate that AA can alleviate depression-like behavior associated with chronic NP.
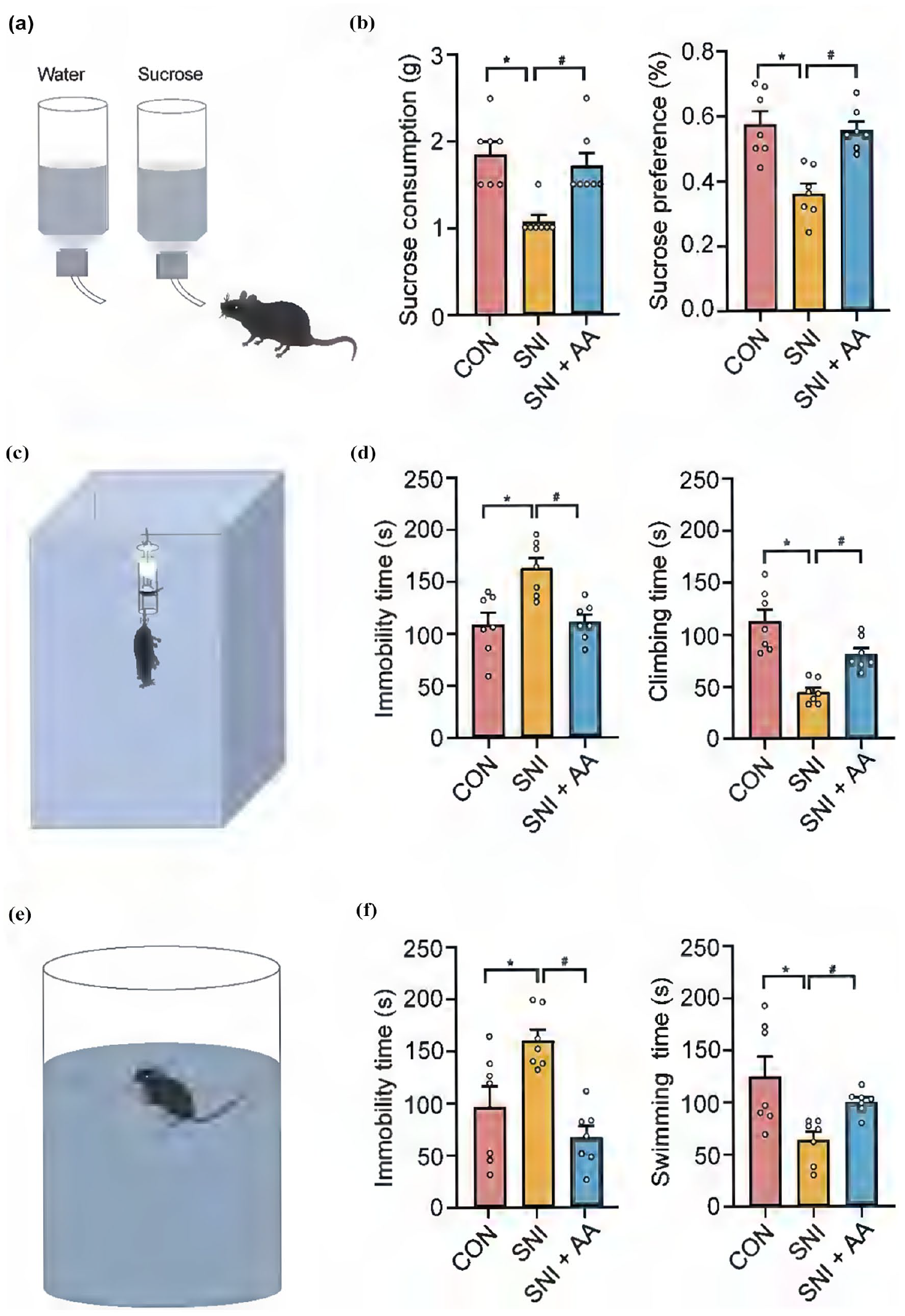
Effects of AA on depression-like behavior in mice with SNI. (a) Schematic diagram of the sucrose preference test. (b) Changes in sucrose consumption (left) and sucrose preference (right) in each group within 2 h after 21 days of AA administration. (c) Schematic diagram of the tail suspension test. (d) Changes in the immobility time (left) and climbing time (right) in the CON, SNI, and SNI + AA groups after 21 days of AA administration. (e) Schematic diagram of the forced swim test. (f) Changes in the immobility time (left) and swimming time (right) in the CON, SNI, and SNI + AA groups after 21 days of AA administration. All data are presented as the mean ± SEM (n = 7). *p < 0.05 compared with the control group; #p < 0.05 compared with the SNI group.
AA mitigated spinal inflammation induced by SNI
NP represents a chronic pain condition that is marked by inflammation. 28 We used HE and immunofluorescence staining to estimate the degree of inflammation in mice with SNI. The results showed that inflammatory cell infiltration was more severe in the SNI group (2.78 ± 0.09) than in the CON group (2.26 ± 0.05; p < 0.05; Figure 3(a), (b)). Furthermore, the intensity of fluorescence for IL-1β in the dorsal horn of mice with SNI was substantially strengthened. However, treatment with AA remarkably decreased the severity of inflammation in mice with SNI (SNI: 2.91 ± 0.61; SNI + AA: 1.46 ± 0.31; p < 0.05; Figure 3(c), (d)). Consistently, western blotting showed that treatment with AA strikingly reduced IL-1β levels of mice with SNI (SNI: 2.52 ± 0.55; SNI + AA: 1.61 ± 0.52; p < 0.05; Figure 3(e), (f)). These data manifest that AA mitigates inflammation by lessening the manifestation of pro-inflammatory factors.

Effects of AA on inflammatory cell infiltration and IL-1β expression in the spinal cord tissues of mice with SNI. (a) Representative images of HE staining of spinal cord tissue sections from mice in the CON, SNI, and SNI + AA groups (scale bar = 20 μm). (b) Quantitative analysis of HE staining scores. (c) Representative images of immunofluorescence staining for IL-1β in the spinal dorsal horns of mice in the three groups. (d) Quantitative analysis of the fluorescence intensity of IL-1β in the three groups (scale bar = 20 μm). (e) Western blotting analysis of IL-1β protein expression levels in the spinal cord tissues of mice in the three groups. (f) Quantification of the relative grayscale values of protein bands. All data are expressed as the mean ± SD (n = 5). *p < 0.05 compared with the control group; #p < 0.05 compared with the SNI group.
AA reduced the activity of NLRP3 inflammasome in the spinal cord of mice with SNI
NLRP3 inflammasome can govern the secretion of the pro-inflammatory cytokine IL-1β by activating caspase-1. 29 To verify the correlation between AA and pro-inflammatory factors, we measured the fluorescence intensity and protein expression of NLRP3 and caspase-1. In contrast to the CON group, the SNI group had higher fluorescence intensities of NLRP3 (relative intensity: 2.89 ± 0.70; Figure 4(a), (b)) and caspase-1 (relative intensity: 2.36 ± 0.47; Figure 4(c), (d)) and higher protein expression levels of NLRP3 (relative gray value: 1.15 ± 0.14) and cleaved caspase-1 (relative gray value: 2.01 ± 0.04; Figure 4(e), (f)) in the dorsal horn. However, treatment with AA reduced the fluorescence intensity and protein expression of NLRP3 and caspase-1 in mice with SNI. These outcomes imply that AA alleviates spinal cord inflammation by impeding the activity of NLRP3 inflammasome and caspase-1.

Effects of AA on NLRP3 inflammasome activation. (a) Representative images of immunofluorescence staining for NLRP3 in the spinal dorsal horns of mice in the CON, SNI, and SNI + AA groups (scale bar = 20 μm). (b) Quantitative analysis of the fluorescence intensity of NLRP3. (c) Representative images of immunofluorescence staining for caspase-1 in the spinal dorsal horns of mice in the three groups (scale bar = 20 μm). (d) Quantitative analysis of the fluorescence intensity of caspase-1. (e) Western blotting analysis of NLRP3, total caspase-1, and cleaved caspase-1 protein expression levels in the spinal cord tissues of mice in the three groups. (f) Quantitative analysis of the relative grayscale values of protein bands. All data are presented as the mean ± SD (n = 5). *p < 0.05 compared with the control group; #p < 0.05 compared with the SNI group.
AA magnified Nrf2 expression and SOD activity in mice with SNI
Acting as a transcription factor, Nrf2 has the ability to regulate the endogenous antioxidant defense mechanism, playing an important role in antioxidant defenses. 30 To assess the antioxidant activity of AA in NP, we evaluated the fluorescence intensity and protein expression of Nrf2. By comparison with the CON group, the SNI group had a significantly lower fluorescence intensity (CON: 1.00 ± 0.52; SNI: 0.33 ± 0.16; p < 0.05; Figure 5(a), (b)) and protein expression level (CON: 0.92 ± 0.08; SNI: 0.55 ± 0.06; p < 0.05; Figure 5(c), (d)) of Nrf2 in the spinal dorsal horn. However, treatment with AA significantly increased the fluorescence intensity (0.96 ± 0.25, p < 0.05; Figure 5(a), (b)) and protein expression level (1.02 ± 0.07; p < 0.05; Figure 5(c), (d)) of Nrf2 in mice with SNI.

Effects of AA on Nrf2 expression and SOD activity in the spinal cord tissues of mice with SNI. (a) Representative images of immunofluorescence staining for Nrf2 in the spinal dorsal horns of mice in the CON, SNI, and SNI + AA groups (scale bar = 20 μm). (b) Quantitative analysis of the fluorescence intensity of Nrf2 in the three groups. (c) Western blotting analysis of Nrf2 protein expression levels in the spinal cord tissues of mice in the three groups. (d) Relative grayscale values of Nrf2 protein expression levels in the three groups. (e) SOD activity in the spinal cord tissues of mice in the three groups. All data are presented as the mean ± SD (n = 5). *p < 0.05 compared with the control group; #p < 0.05 compared with the SNI group.
SOD is an important antioxidant regulated by Nrf2, and its expression can reflect the antioxidant function of Nrf2. 31 We measured SOD activity to assess the effects of AA on oxidative stress. The SNI group showed significantly lower SOD activity as compared with the CON group (CON: 15.20 ± 0.30 U/mg; SNI: 9.65 ± 1.11 U/mg; p < 0.05). However, treatment with AA significantly increased SOD activity in mice with SNI (SNI: 9.65 ± 1.11 U/mg; SNI + AA: 14.00 ± 1.02 U/mg; p < 0.05; Figure 5(e)). These determinations show that AA exerts its anti-inflammatory impacts by initiating the Nrf2-governed antioxidant pathway.
AA activated AMPK signaling in the spinal cord of mice with SNI
AMPK functions as a redox sensor, and its activation can alleviate diseases associated with oxidative stress. 32 To determine the pathological mechanisms underlying AA, we performed molecular docking between AMPK and AA. Data showed that AA and AMPK formed four valence bonds at SER-225 and ALA-226 residues, with a binding affinity of 5.0 kcal/mol (Figure 6(a)–(c)). In contrast to the CON group, the SNI group showed a notable decrease in the fluorescence intensity of P-AMPK in the spinal dorsal horn (CON: 1.17 ± 0.15; SNI: 0.45 ± 0.16; p < 0.05). However, mice with SNI exhibited an appreciable increase in the fluorescence intensity of P-AMPK after treatment with AA (SNI: 0.45 ± 0.16; SNI + AA: 1.05 ± 0.24; p < 0.05; Figure 6(c), (d)). Western blotting showed that P-AMPK/AMPK expression was lower in the SNI group (0.56 ± 0.32) than in the CON group (1.00 ± 0.19; p < 0.05; Figure 6(e), (f)). Nonetheless, treatment with AA led to an elevation in the expression of P-AMPK/AMPK (1.14 ± 0.10) in mice with SNI (p < 0.05; Figure 6(e), (f)). These results imply that AA generates anti-inflammatory effects through actuating the AMPK signaling pathway.

Effects of AA on AMPK expression and activity. (a) Molecular docking of AA with AMPK. (b) Enlarged image of the binding site. (c) Interaction bond between AMPK and AA. The AMPK protein is shown in blue, whereas AA is shown in green. The interacting residues are represented in red, the bonds are represented as yellow dashed lines, and bond lengths are represented as numbers. (d) Representative images of immunofluorescence staining for P-AMPK in the spinal dorsal horns of mice in the CON, SNI, and SNI + AA groups. (e) Quantitative analysis of the fluorescence intensity of P-AMPK in the three groups. (f) Western blotting analysis of P-AMPK (Thr172) expression levels in the spinal cord tissues of mice in the three groups. (g) Relative grayscale values of P-AMPK expression levels in the three groups. All data are presented as the mean ± SD (n = 5). *p < 0.05 compared with the control group; #p < 0.05 compared with the SNI group.
AA treatment restored the energy of LFPs in the spinal cord of mice with SNI
LFPs are electrical signals produced by the activity of neuron groups, reflecting both the interactions among neurons and the overall activity of the neural network. 33 Although AA is famed for its antioxidant properties and potential neuroprotective effects, its specific role in modulating spinal cord oscillations remains unclear. To investigate the impact of AA on spinal cord LFPs in mice with NP, we recorded LFPs and assessed the power spectral density. The neural oscillations were examined in the following ranges of frequencies: theta (4–8 Hz), alpha (8–13 Hz), and beta (13–30 Hz; Figure 7(a)). The frequency spectrogram demonstrated a notable elevation in theta, alpha, and beta band powers in the SNI group (Figure 7(b), (c)). Statistical analysis further revealed a marked increase in theta (CON: 0.06 ± 0.01; SNI: 5.50 ± 0.32; p < 0.01), alpha (CON: 0.04 ± 0.01; SNI: 0.58 ± 0.05; p < 0.01), and beta (CON: 0.06 ± 0.01; SNI: 0.54 ± 0.04; p < 0.01) band powers in mice with SNI (Figure 7(d)). Nonetheless, treatment with AA effectively counteracted the elevation in the theta (SNI: 5.50 ± 0.32; SNI + AA: 0.05 ± 0.01; p < 0.01), alpha (SNI: 0.58 ± 0.05; SNI + AA: 0.04 ± 0.01; p < 0.01), and beta (SNI: 0.54 ± 0.04; SNI + AA: 0.07 ± 0.02; p < 0.01) band powers in mice with SNI (Figure 7(d)), suggesting the effectiveness of AA in regulating abnormal neural electrophysiological activities. Altogether, these data show that AA alleviates NP by decreasing the activity of neurons.

Treatment with AA restored the energy of local field potentials in the spinal cord tissues of mice with SNI. (a) Time course of raw LFPs in the spinal cord (top, black) and theta, alpha, and beta band-filtered LFPs (blue) in the spinal dorsal horn. (b) Spectrograms of the CON, SNI, and SNI + AA groups. (c) Average LFP power in the CON, SNI, and SNI + AA groups. (d) Statistical analysis of LFP power at different frequency bands. Error bars indicate the mean ± SEM (n = 6 mice). *p < 0.05 compared with the control group; #p < 0.05 compared with the SNI group.
Discussion
In this study, mice with SNI-induced NP were found to have elevated inflammatory responses and oxidative stress levels in the spinal cord. Treatment with AA alleviated NP and associated depression-like behavior in these mice by activating spinal AMPK signaling, inhibiting NLRP3-mediated inflammatory responses, and enhancing Nrf2-mediated antioxidant responses (Figure 8). We will increase the groupings in the future experiments to obtain more comprehensive data, utilizing network analysis to further elucidate the concrete mechanism of AA, and promoting our ability on systematic statistical analysis. AMPK is a protein sensitive to metabolic stress, and its phosphorylation levels are known to decrease under pathological inflammatory conditions. 34 In studies on rodents with peripheral nerve traumatic injury, AMPK has been considered a key and unique target for alleviating NP. 35 Activation of threonine 172 (Thr172) within the phosphorylation kinase domain is essential for achieving maximum AMPK activity, with phosphorylation at this site stimulating AMPK activity by more than 100 times.36,37 Studies on inflammatory pain have demonstrated that both emodin and humic acid exert analgesic effects by activating AMPK phosphorylation at Thr172.38,39 Research conducted on the A study on the SNI model demonstrated that heat shock protein 22 (hsp22) promoted the activation of spinal AMPK by phosphorylating it at Thr172, contributing to relief in NP. Additionally, AMPKα1-knockout mice exhibited increased mechanical pain sensitivity owing to the elevated activity of spinal glutamatergic synapses and production of reactive oxygen species (ROS). 40 Furthermore, AA can sensitize pancreatic cancer cells to erastin-elicited ferroptosis through the AMPK signaling pathway while markedly reducing the toxic effects of erastin on normal cells. 14 The SNI model induces both NP and associated depression, 41 highlighting the close relationship between the AMPK signaling pathway and NP. In this research, we observed a binding affinity of 5.0 kcal/mol between AA and AMPK, with stable docking. Therefore, we speculate that AA alleviates NP and depression-like behavior by binding to AMPK and enhancing its activity.

Schematic diagram of the potential mechanism of action of AA in alleviating neuropathic pain and associated depression-like behavior.
AMPK can mitigate inflammation through various pathways. In ulcerative colitis, GPA peptide can suppress the activation process of NLRP3 inflammasome by enhancing AMPK phosphorylation. 42 In a study on traumatic brain injury, AMPK phosphorylation at Thr172 was found to be suppressed in mouse models, leading to an increase in NLRP3 inflammasome activity. Correspondingly, knockout of AMPK exacerbated traumatic brain injury in the mice. After 24 h, the expression levels of various inflammatory markers, specifically NLRP3, IL-1β, and caspase-1 demonstrated a notable invrease in the knockout group as compared with in the non-knockout group. 43 In spinal cord injury, ulinastatin has been shown to impede the activation of NLRP3 inflammasome by activating the AMPK signaling pathway, thereby attenuating the inflammatory response. 44 Additionally, AMPK add a phosphate groupto the Ser550 site of Nrf2. This phosphorylation, along with the inhibition of GSK3β mediated by AMPK, encourages Nrf2 to accumulate in the nucleus, thereby facilitating the transactivation of genes driven by the antioxidant response element (ARE). 45 The ability of AMPK to mitigate neuroinflammation by inhibiting NLRP3 inflammasome activity has been established in multiple studies. 42 AA increases the ratio of P-AMPK to AMPK through multiple pathways, including ROS-mediated signal regulation, 46 the activation of downstream effector molecules (such as PGC-1α/SIRT1 47 ), and the reprogramming of energy metabolism. 48 Additionally, while this research has identified AMPK signaling pathway as a key pathway, the exploration of this pathway remains insufficient. The current conclusions regarding the relationship between AA and AMPK signaling pathway have certain limitations. Future research will focus on the specific molecular interactions and signaling cascades related to NP and their direct effects on AMPK signaling pathway, with more detailed and in-depth studies.
In conditions of oxidative stress, Nrf2 separates from Keap1 in the cytoplasm and moves to the nucleus. In the nucleus, Nrf2 forms heterodimers with Maf, enhancing their affinity for the antioxidant response element (ARE) and facilitating binding. This interaction promotes the transcription of various antioxidant enzymes, resulting in antioxidant and analgesic effects. 49 Prolonged inflammation may lead to the onset of depression, as excessive production of inflammatory factors can contribute to the development and exacerbation of mental illness. 50 The NLRP3 pathway is intricately linked with the worsening of depression. 51 NLRP3 inflammasome gets activated in those afflicted with depression, and the absence of NLRP3 has been shown to alleviate depression-like behavior and inhibit inflammatory responses in mice. 52 In this study, we found that AA alleviates NP and associated depression by activating AMPK, increasing the expression of Nrf2, and preventing the NLRP3 inflammasome from being activated.
LFPs are critical indicators of nociceptive processing and pain modulation within the spinal cord. 53 They are primarily evoked by the activation of primary afferent fibers, particularly C-fibers, which are responsible for transmitting pain signals. In this study, we found increased energy of low-frequency oscillations of mice with SNI, which is consistent with the findings of an earlier research.54,55 The energy of these low-frequency oscillations was restored after intraperitoneal injection of AA. These research outcomes yield valuable insights into the mechanisms responsible for pain perception, offering novel avenues for therapeutic intervention. Altogether, this study suggests that AA inhibits spinal cord inflammation and alleviates depression-like behavior by activating the AMPK-Nrf2 pathway.
Footnotes
Author contributions
Lixin Yao, Mengwei Zhang, Shuang Wang, and Qing Yao conducted experiments and data analysis. Lingli Qiao, Shaohui Chen, Zhongli Qin, Wei Meng, Min Xie, Haili Zhu, and Ling Liu made significant contributions to study design, engaged in in-depth discussions on the results, and participated in manuscript writing. All the authors have reviewed and consented to the manuscript content.
Availability of data and materials
The data arising from this study will be made available by the corresponding author in response to a reasonable request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the National Natural Science Foundation of China (No: 32100823), Hubei Provincial Natural Science Foundation of China (No: 2023AFB1087, and 2024AFB1029), and Hubei Provincial Natural Science Foundation and innovation joint of China (No: 2023AFD142).
Ethics approval and consent to participate
All animal experiments were approved by the Ethics Committee of Hubei University of Science and Technology, Xianning, China (SYXK2023-0071).
Consent for publication
All of the authors endorsed the idea of its publication.