
Abstract
Neuropathic pain (NP) is often accompanied by psychiatric comorbidities and currently lacks effective treatment. Prior research has shown that HDAC6 plays a crucial role in pain sensitization, but the specific mechanisms remain unclear. HDAC6 inhibitors have been found to alleviate mechanical allodynia caused by inflammation and peripheral nerve damage. In this study, we investigated the cellular mechanisms of HDAC6 in the development and maintenance of neuropathic pain. Our findings indicate that HDAC6 expression in the spinal cord (SC) is upregulated in a time-dependent manner following chronic constriction injury (CCI). HDAC6 is primarily expressed in neurons and microglia in the spinal cord. CCI-induced HDAC6 production was abolished by intrathecal injection of a microglia inhibitor. ACY-1215, a specific HDAC6 inhibitor, significantly reduced CCI-induced mechanical allodynia, but not thermal hyperalgesia. ACY-1215 also inhibited neuron activation and suppressed CCI-induced pyroptosis and neuroinflammatory responses. In summary, our results suggest that HDAC6 contributes to the development and maintenance of NP through neuronal activation and neuroinflammation. HDAC6 may be a promising target for treating NP.
Introduction
Neuropathic pain (NP) refers to an abnormal sensation of pain caused by damage or injury to the somatosensory nervous system. This condition is characterized by symptoms of allodynia and hyperalgesia, which can significantly impact a patient's quality of life and pose a burden on society both clinically and economically. 1 Unfortunately, current interventions for NP do not provide sufficient pain relief, making it an urgent priority to understand the underlying pathophysiological mechanisms of NP to improve treatments.
Previous studies have shown that central sensitization and neuroinflammation contribute to the development of NP by enhancing synaptic efficacy and increasing neuronal excitability in nociceptive pathways. 2 Similar findings have been confirmed in spinal dorsal horn neuronal circuits in patients with NP and inflammatory pain. 3 Nevertheless, the underlying mechanism remains fundamentally obscure.
Histone acetylation in the spinal cord has recently been implicated in nociceptive sensitization in animal models of neuropathic pain. Research has shown 4 that HDACs are increased in the spinal cord of chronic constriction injury (CCI) mice, while the acetylation level of H3 histones is decreased. Additionally, the application of non-specific HDAC inhibitors can alleviate pain behaviors in neuropathic pain mice. Therefore, modulating the activity of HDACs may potentially reduce the expression of genes related to pain sensitization, thus alleviating neuropathic pain.
HDAC6 is a class IIb histone deacetylase that primarily deacetylates non-histone proteins, such as tubulin and heat shock proteins, that play a role in synaptic plasticity. 5 The suppression of HDAC6 activity has been suggested as a potential therapeutic approach for neurodegenerative diseases and also shows potential for cancer treatment.6,7 Recent studies have demonstrated the significant involvement of HDAC6 in the regulation of pain perception. The administration of an HDAC6 inhibitor has been found to alleviate chemotherapy-induced heightened sensitivity to mechanical stimuli 8 and reduce nerve injury and inflammation caused by SNI and CFA. 9
The NOD-like receptor (NLR) family pyrin domain containing protein 3 (NLRP3) inflammasome is a crucial molecular platform, 10 which can be triggered by internal and external harmful signals to contribute to the release of IL-18 and IL-1β and facilitate their maturation.11,12 At the same time, the activated inflammasome initiates pyroptosis, a recently discovered proinflammatory programmed cell death, which causes the cleavage of the cell membrane and induces the leakage of cellular contents, resulting in inflammatory cascades. 13 Several studies have indicated that the activation of NLRP3 inflammasome plays a role in the development of neuropathic pain.14,15 In the meantime, pyroptosis plays a role in the neuroinflammation caused by Alzheimer’s disease 16 or intracerebral hemorrhage. 17 Therefore, regulating pyroptosis through inflammasome-mediated pathways is a promising strategy for treating neuropathic pain. While HDAC6 has been shown to play a crucial role in pyroptosis,18,19 it’s involvement in the pathogenesis of NP through pyroptosis has not been previously reported.
In this study, we observed that HDAC6 was upregulated in the spinal cord of CCI mice and was distributed in neurons and microglia of the spinal dorsal horn. Given the critical role of HDAC6 in inflammation related to macrophages, we investigated the relationship between HDAC6 and microglial activation in the spinal cord. Our results showed that microglial activation induced the expression of HDAC6. Furthermore, blocking HDAC6 suppressed neuronal activation and reversed CCI-induced pyroptosis through the NF-κB/NLRP3 pathway. Additionally, it relieved neuroinflammation by suppressing TNF-α and IL-6 in the spinal cord. These findings suggest that targeting HDAC6 may be a potential therapeutic option for treating chronic NP.
Materials and methods
Animals
Male C57BL/6 mice (8 weeks old, weighing 21–25 g) were procured from Xuzhou Medical University (SYXK 2016-0028). The mice were housed in standard conditions, with four to six mice per cage, in a room maintained at 22 ± 2°C and a 12 h/12 h dark/light cycle (08:00 a.m.–08:00 p.m.). They were provided with ad libitum access to food and water. Prior to the experiments, the mice were acclimatized for 7 days and kept in group housing with the same cage mates throughout the acclimation and experiment. All animal experiments were conducted in accordance with the Use of Laboratory Animals and the Animal Ethics Committee of Xuzhou Medical University, Jiangsu, China. The experiments were performed in compliance with the Animal Research Reporting In Vivo Experiments (ARRIVE) guidelines.
Neuropathic pain model
A neuropathic pain model, known as the CCI model, was established by loosely ligating the sciatic nerve, following a previously described method. 20 In brief, mice were anesthetized using a xylazine-ketamine mixture (5–10/50–100 mg/kg, i.p.). An incision was made on the lateral surface of the left thigh (opposite to the site of viral injection) and a section was created through the biceps femoris and gluteus muscles. These muscles were separated through blunt dissection, exposing the sciatic nerve, which was then gently retracted. Three loose ligations were applied to the dorsal third to half of the common sciatic nerve at the upper-thigh level using nonabsorbent 5–0 silk suture (Ethicon).
Pain behavioral quantification
The testing procedure was conducted in accordance with previously established protocols 21 and our published report. 22 To assess thermal hyperalgesia, we measured the paw withdrawal latency (PWL) in response to radiant heat stimulation. A Plantar Analgesia Meter (IITC Life Science Inc., CA, USA) served as the source of radiant heat. Mechanical allodynia was evaluated by measuring the paw withdrawal threshold (PWT) in response to stimulation with Von Frey hair (Stoelting, Wood Dale, IL, USA). The protocol employed was similar to Dixon’s up and down method. 23 The behavioral testing was carried out by an investigator who was blinded to the treatment.
Drugs and administration
Minocycline was dissolved in PBS to prepare for intrathecal injection. The intrathecal injections were administered using a 10-μl microinjection syringe, following the previously described method. 24 The syringe was carefully inserted into the intervertebral space between the lumbar 5 (L5) and 6 (L6) regions of the spinal cord in a conscious mouse. The accuracy of each injection was assessed by observing a reflexive flick of the tail. For the intrathecal injections after CCI from day 7 to day 10, a dosage of 25 mg/kg was used. To inhibit HDAC6, ACY-1215 (HY-16026, MCE, China), a specific inhibitor, was dissolved in a solution containing 10% dimethyl sulfoxide (DMSO) and 40% polyethylene glycol 400 (HY–Y0873 A, MCE, China) in saline. This inhibitor was intraperitoneally injected at a dosage of 25 mg/kg once a day for 14 consecutive days, starting from day 1 to day 14 after the operation. 25 As a control, an equal volume of vehicle was intraperitoneally injected instead of ACY-1215, serving as a sham.
Western blot analysis
The L4-5 spinal cords were swiftly extracted from deeply anesthetized mice, followed by dissection and preservation in liquid nitrogen. The tissue samples were homogenized in a lysis buffer (Bio-Rad Laboratories, Hercules, California, United States) containing a mixture of protease and phosphatase inhibitors (Sigma Aldrich, St Louis, United States). After centrifugation at 12000 g for 15 min at 4°C, the supernatants were collected and the protein concentration was determined using the bicinchoninic acid assay. To ensure equal protein content among samples, the total protein was standardized. Subsequently, the samples were heated at 100°C for 5 min in a loading buffer (2% sodium dodecyl sulfate, 100 mM dithiothreitol, 10% glycerol, and 0.02% bromophenol blue) to induce dissociation. Equivalent amounts of protein (80 μg) were then separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride membrane (PVDF, Millipore, Billerica, Massachusetts, United States). The membranes were immersed in a blocking solution, consisting of Tris-buffered saline with 0.02% Tween (TBST) and 5% non-fat dry milk, for 2 h at room temperature. Subsequently, they were incubated overnight at 4°C with the following primary antibodies: rabbit anti-HDAC6 polyclonal antibody (1:1000, NBP1-45611, NOVUS Biotechnology, United States), rabbit anti-NLRP3 (1:1000, ET1610-93, Huabio, China), NF-κB p65 (1:1000, 8242S, CST, United States), caspase-1 p20 (1:500, ER1905-47, Huabio, China), IL-18 polyclonal antibody (1:5000, 10663-1-AP, proteintech, United States), and β-actin (1:10000, ab8226, Abcam, United States).
Immunofluorescence
Under deep anesthesia, the L4-5 spinal segment of the mice was delicately dissected and post-fixed in a 4% PFA solution for a duration of 3 h. It was then carefully transferred into Tris-buffered saline (TBS) enriched with 30% sucrose, ensuring its preservation at a cool temperature of 4°C until further use. The spinal sections, sliced with utmost precision to a thickness of 30 μm, were meticulously stored in TBS, ensuring their optimal condition. The tissue sections were initially subjected to blocking with 5% donkey serum in 0.3% Triton X-100 for a duration of 1 h at room temperature, then incubated overnight at 4°C with the following primary antibodies: rabbit anti-HDAC6 polyclonal antibody ((1:200, NBP1-45611, NOVUS Biotechnology, United States)), goat anti-IBA1 polyclonal antibody (1:400, ab5076, Abcam, Cambridge, Massachusetts, United States), rabbit anti-p-CREB monoclonal antibody (1:800, Alexa #9198, Cell Signaling Technology, Beverly, Massachusetts, United States), rabbit anti-c-Fos antibody (1:1000, ab208942, Abcam, Cambridge, Massachusetts, United States). The sections were then washed 3 times with PBS and incubated with the specific secondary antibodies raised in donkey serum(conjugated to Alexa Fluor 488 or 594, Invitrogen, Carlsbad, California, United States). Subsequently, the sections were incubated overnight at a temperature of 4°C.
For double staining, sections were incubated again in the same blocking solution and the staining process was performed once again. After immunostaining procedures, the sections were examined using a laser scanning confocal microscopy (FV1000, Olympus, Tokyo, Japan).
Statistical analysis
GraphPad Prism 9.5 (GraphPad Software, La Jolla, California, United States) was used to conduct all statistical analyses. The data were presented as mean ± SEM. The results from the western blot, Immunofluorescence and ELISA study were tested using repeated measures of one-way analysis of variance (ANOVA), followed by the Bonferroni post-hoc test to evaluate the difference between each group. Behavioral data were analyzed using two-way repeated measures ANOVA with post-hoc Bonferroni test was employed to analyze data with multiple time points. The threshold for statistical significance in all analyses was set at p < .05. The data analysis was conducted by an investigator who was blinded to the experimental design.
Results
Expression and distribution of HDAC6 in spinal cord after CCI
To examined whether HDAC6 could be upregulated under NP state, we first detected the expression and distribution of HDAC6 in the SC after CCI. The western blot analysis showed that CCI induced a rapid-onset and long-lasting increased HDAC6 expression 3 days after CCI and lasted for more than 21 days Figure 1(a). To investigate the cell distribution of HDAC6 in DH following CCI treatment, we performed double immunostaining of HDAC6 with three major spinal nerve cell-specific markers: NeuN (for neurons), GFAP (for astrocytes), and IBA1 (for microglia). Confocal images showed that the double immunostaining of HDAC6 was extensively colocalized with NeuN and IBA1 but not GFAP, suggesting that HDAC6 is expressed in neurons and microglias, but not astrocytes in CCI mice Figure 1(b). These results demonstrate that spinal HDAC6 is upregulated after CCI, and predominately expressed in neurons and microglia. Expression and distribution of HDAC6 in spinal cord after CCI. (a) Western blot analysis showed the time course of HDAC6 expression in sham and CCI mice. Representative bands were shown on the top, data summary was shown on the bottom. *p < .05, **p < .01 versus sham group; one-way ANOVA with post-hoc Bonferroni test, n = 4 for each group. (b) Double staining showed that HDAC6 (green) is colocalized with NeuN, a marker for neurons and IBA1, a marker for microglia, but not with GFAP, a marker for astrocytes. Tissues were collected 10 days after CCI. Two single-stained images were merged. Original magnification: 200 × for all the confocal images showing the dorsal horn ipsilateral to CCI (bar = 100 μm).
The increased expression of HDAC6 induced by CCI is mediated by microglial activation
Microglia is considered the innate immune cells of the central nervous system. Upon activation, microglia release pro-inflammatory cytokines and cytotoxic compounds that disrupt homeostatic processes and neuronal functions. It has been demonstrated that HDAC6 plays an important role in macrophages mediated inflammation. Here we investigated the relationship between spinal microglia and HDAC6. Minocycline, an inhibitor of microglia was used to suppress microglial activation. As expected, minocycline decreased the expression of HDAC6 in SC after CCI (Figure 2). These results suggest that microglial activation mediates HDAC6 hyperexpression in SC after CCI. Intrathecal administration of minocycline reversed CCI-induced upregulation of HDAC6 in the spinal cord. (a) Inhibitory effect of minocycline on CCI-induced increased expression of HDAC6. **p < .01 versus sham group, #p < .05 versus CCI + vehicle group; one-way ANOVA with post-hoc Bonferroni test, n = 5 for each group. (b)-(c) Representative confocal images and statistical graph (bar = 100 μm). ****p < .0001 versus sham group, ###p < .001 versus CCI + vehicle group, one-way ANOVA with post-hoc Bonferroni test, n = 12 neurons from three mice for each group.
Inhibition of HDAC6 alleviates mechanical allodynia, neuronal activation in the CCI model of peripheral nerve injury
To examine the role of HDAC6 in NP modulation, we used ACY-1215, an inhibitor of HDAC6 to suppress HDAC6. We tested the pain behaviors in mice. As shown, CCI mice treated with ACY-1215 (25 mg/kg) showed a recovery in the decreased levels of mechanical threshold at day 5∼14 after ACY-1215 administration compared with vehicle treatment, but ACY-1215 did not alleviate nociceptive responses in the hot plate assay. These data suggest that ACY-1215 modulates sensory hypersensitivity, but it does not impact acute thermal nociceptive thresholds.
Previous studies have shown that neurochemical alterations such as c-Fos, p-CREB in the ipsilateral DH induced by CCI result in behavioral changes of NP.26,27 Here we investigated whether HDAC6 is involved in the activation of neurons (c-Fos and p-CREB) caused by CCI. Immunofluorescence results showed that the increased expressions of c-Fos and p-CREB induced by CCI were greatly suppressed by ACY-1215 (25 mg/kg, i.p., once a day from day 1 to 14 after CCI) for 14 consecutive days. The results indicate that spinal HDAC6 may directly mediate the activation of neurons under NP condition (Figure 3). Inhibition of HDAC6 suppressed spinal cells pyroptosis and neuroinflammation induced by CCI. (a)–(b) Inhibitory effect of ACY-1215 on CCI-induced increased expressions of NLRP3, NF-κB p65, caspase-1 p20 and IL-18. **p < .01 versus sham, ##p < .01, #p < .05 versus CCI + vehicle group; one-way ANOVA with post-hoc Bonferroni test, n = 5 for each group. (c)–(d) CCI-induced increased expressions of TNF-α and IL-6 were suppressed by HDAC6 inhibitor ACY-1215 at day 14 after CCI. ***p < .001 versus sham. ###p < .001 versus CCI + vehicle group; one-way ANOVA with post-hoc Bonferroni test, n = 4 for each group.
Inhibition of HDAC6 suppresses pyroptosis and neuroinflammation via NF-κB/NLRP3 pathway in spinal cells
Pyroptosis is involved in the formation and progression of pain development.28,29 Recent studies have demonstrated that HDAC6 regulates autophagy and NLRP3 inflammasome activation through various mechanisms. The NLRP3 inflammasome promotes the onset of cell pyroptosis. The NF-κB signaling pathway usually activates transcription of the NLRP3 inflammasome. One previous study has shown that CFA-induced inflammatory pain increased the level of the NLRP3 inflammasome in the spinal dorsal horn. 28 The pyroptosis-associated proteins are upregulated during CCI that can induce NP,30,31 but whether HDAC6 is involved in CCI-induced pyroptosis is largely unknown. Here, we investigated the potential role of HDAC6 in pyroptosis of SC cells. The results showed that blocking of HDAC6 by ACY-1215 reversed the increased expressions of NLRP3, NF-κB p65, caspase-1 p20 and IL-18 in SC after CCI treatment. Our data suggest that HDAC6 may mediate pyroptosis by activating NF-κB/NLRP3 pathway in CCI-induced NP.
As known, spinal microglial activation mediated neuroinflammation is crucial for pain development. Inhibition of HDAC6 suppress microglia activation (Figure 4), thus here we investigated the effect of HDAC6 suppression on neuroinflammation. ELISA experiments showed the expression of proinflammatory cytokines, including TNF-α and IL-6, were upregulated after CCI. HDAC6 inhibitor reversed the levels of TNF-α and IL-6. These results suggest that HDAC6 plays an important role in CCI induced neuroinflammation. Intrathecal administration of ACY-1215 reversed CCI-induced upregulation of HDAC6, pain hyperalgesia and neuronal activation in the spinal cord. (a) Schematic timeline for drug administration, behavioral detection, and tissue extraction. (b)–(c) ACY-1215 (25 mg/kg), an inhibitor of HDAC6 reversed CCI-induced mechanical allodynia, but not thermal hyperalgesia in the feet ipsilateral. *p < .05, **p < .01 versus sham, #p < .05, ##p < .01 versus CCI + vehicle; two-way ANOVA with repeated measures followed by Bonferroni post-tests, n = 7 for each group. (d) Representative immunofluorescence images of spinal dorsal horn showing c-Fos+ or p-CREB+ cells in sham, CCI, CCI + vehicle and CCI + ACY-1215 groups (bar = 100 μm). (e)-(f) Statistical graph showing the immunofluorescence intensity of c-Fos+ or p-CREB+ in sham, CCI, CCI + vehicle, and CCI + ACY-1215 groups. Tissues were collected 4 h after the last intraperitoneally injection. **p < .01 versus sham, ##p < .01versus CCI + vehicle; one-way ANOVA with post-hoc Bonferroni test, n = 15 neurons from five mice for each group.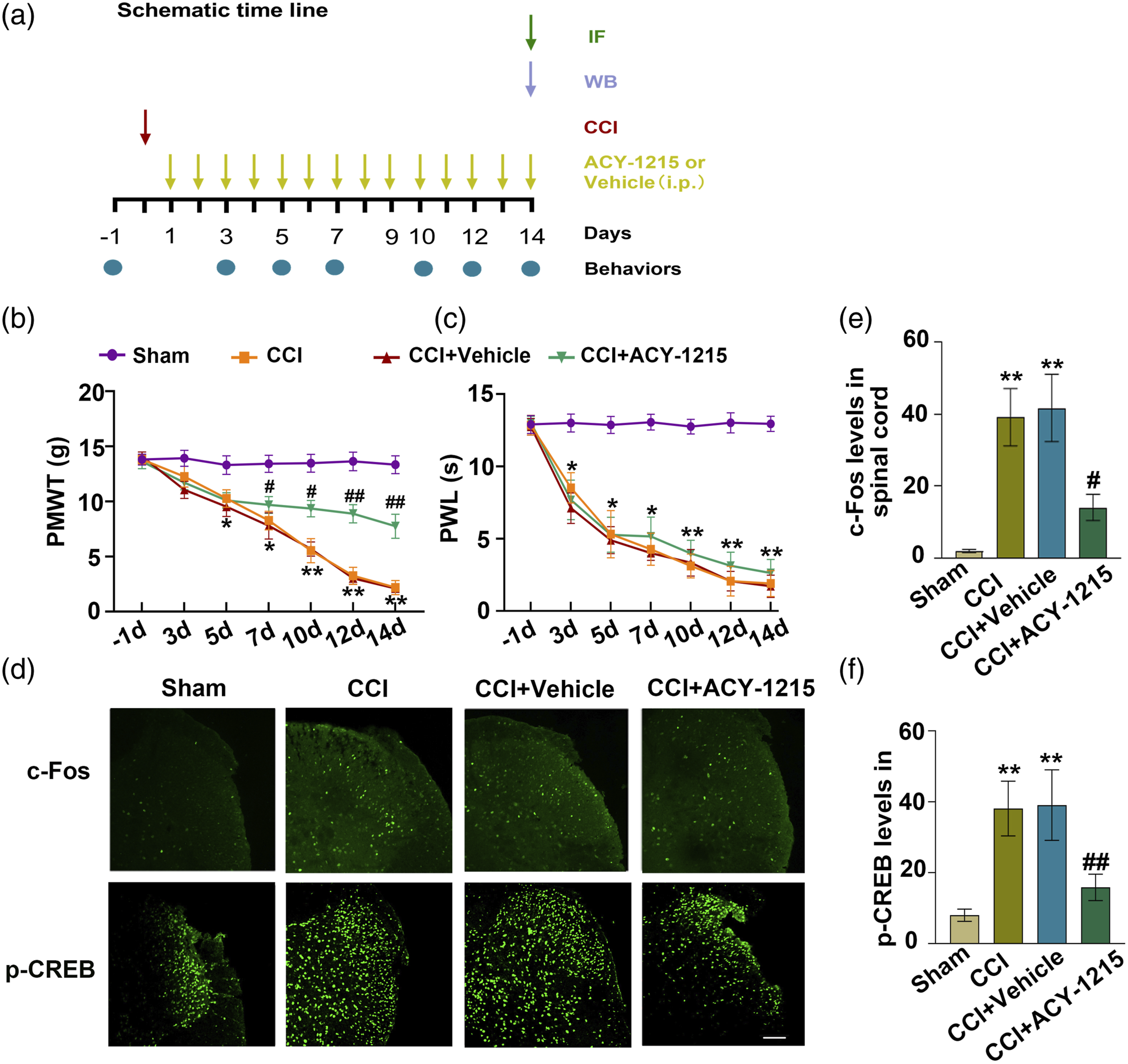
Discussion
Dysregulated activity and expression of HDAC6 have been implicated in the pathogenesis of various human diseases, including lung cancer and cognitive disorders.32–34 In our study, we investigated the potential of ACY-1215, a specific inhibitor of HDAC6 deacetylation activity, as a novel therapeutic strategy for NP. Our results showed that ACY-1215 treatment effectively suppressed CCI-induced neuronal activation, as evidenced by reduced expression of c-Fos. These effects were consistent with the inhibitory effects of HDAC6 on synaptic plasticity.35,36 Furthermore, ACY-1215 administration reversed CCI-induced microglial activation, cell pyroptosis, and pro-inflammatory cytokine production. This study provides the first evidence that HDAC6 may serve as a promising therapeutic target for NP.
Growing evidence suggests that glial activation and neuronal activation in the spinal cord (SC) play crucial roles in the development and maintenance of pain hypersensitivity in NP.37–39 HDACs have been implicated in neuronal activation and/or glial activation in the SC,40,41 and some of them contribute to NP processing. 42 HDAC6, in particular, has been shown to be expressed in key locations of the pain transmission pathway in animal models of pathological pain.9,25
In our study, we investigated the expression, distribution, and function of HDAC6 in the CCI model of NP. Since neuronal activation is crucial in regulating pain, the involvement of HDAC6 in neuronal activation and neuronal plasticity regulation has been established.5,6 HDAC6 is exclusively localized in the cytoplasm and is highly expressed in the hippocampus, where it regulates NP-induced cognitive impairment by modulating neuronal activation. 36 Additionally, HDAC6 mutants display a limited hindrance to homeostatic plasticity, indicating that HDAC6 might be necessary for maintaining synaptic strength. 35 In our study, we discovered that HDAC6 is present in dorsal neurons of the SC. Inhibiting HDAC6 suppresses the expression of c-Fos and p-CREB, suggesting that HDAC6 may regulate NP by modulating neuronal activation in the SC.
Prior research has indicated that HDAC6 plays a pivotal role in regulating the modulation of inflammation.18,19 The use of TBA, a pharmacological inhibitor of HDAC6, has been shown to mitigate NLRP3-induced inflammation and provide protection for dopaminergic neurons, potentially through the acetylation of Prx2. 43 A previous investigation discovered that the dynein, an adaptor for HDAC6, is indispensable for the transportation and assembly of NLRP3-associated inflammasomes, both in laboratory settings and in mice. 44 Moreover, our findings demonstrate that HDAC6 is also present in microglia and is induced by the activation of these cells. The inhibition of microglia using minocycline resulted in a reduction in the expression of HDAC6 in the SC.
Given the inhibitory function of HDAC6 on gene expression, the current investigation revealed that CCI elicited distinct mechanisms in promoting the transcription of pro-inflammatory cytokines and the expression of microglial IBA1. c-Fos, a crucial component involved in neuronal activation, has been extensively documented to be implicated in the release of various chemokines by activated microglia, including neurotrophic factors that impact neuronal viability and pro-inflammatory agents.37,45 Previous research has elucidated the transmission of signals from activated microglia to neurons, such as TNF-α and IL-1β. All the findings provided substantiation that microglial activation detrimentally influenced neuronal transcription.
The NLRP3 inflammasome is a multimeric protein complex that has been implicated in the onset and maintenance of neuropathic pain (NP). However, the molecular mechanisms involving upstream and downstream effectors of NP remain to be elucidated.46,47 Pyroptosis, a unique inflammatory regulated cell death mechanism, has been reported to be involved in various chronic inflammatory diseases and is dependent on caspase-1 activity. Nuclear factor-kappa B (NF-κB), a transcription factor, plays a pivotal role in the initiation of inflammation. During chronic inflammation, the NLRP3 inflammasome activates NF-κB signaling, leading to increased production of inflammatory mediators. HDAC6 serves as a pivotal controller of NLRP3, and its inhibition attenuates NLRP3 inflammasome activation in LPS-induced mice. 48 In our study, we found that CCI promoted pyroptosis in spinal cord neurons of mice, as evidenced by increased expression of cleaved Caspase1, NLRP3, and IL-18 release. Inhibition of HDAC6 suppressed CCI-induced pyroptosis, which was partly mediated by p65 acetylation and NLRP3 transcription. Therefore, targeting the HDAC6/NF-κB/NLRP3/pyroptosis signaling pathway to prevent NP is an attractive clinical application research.
Our investigation has revealed a previously unrecognized cell-specific role of HDAC6 in regulating the transcriptional activity of spinal neurons and the interaction between neurons and microglia. Further investigations are needed to determine which types of receptors on neurons are affected by neuroinflammation and which pathways and potential transmitter-receptor interactions regulate glial activation.
In summary, our findings consistently demonstrated that microglial activation increased HDAC6 expression leading to activation of neurons in SC, which resulted in NP. Repression of HDAC6 rescued microglial and neuronal activation and suppressed pyroptosis in the SC, ultimately preventing neuropathic pain in mice.
Footnotes
Acknowledgements
We thank all participants who contributed to this study. We express our sincere appreciation to the reviewers for their constructive comments.
Author contributions
CG and Z-PW as the primary and corresponding authors, overseeing all experiments and providing funding for the study. KS and TZ were responsible for drafting the manuscript and planning the research. HZ and J-RH conducted the experiments and analyzed the data. NS, KS and HZ made revisions to the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China [grant numbers 82171199, 82001169].