
Abstract
The opioid crisis has highlighted the urgent need for alternative pain management strategies. This review explores novel non-opioid targets and pathways involved in pain modulation, highlighting advancements in understanding and therapeutic potential. Pain, a multifaceted phenomenon with nociceptive, neuropathic, and inflammatory components, involves intricate molecular signaling cascades. Key pathways reviewed include voltage-gated sodium channels (Nav1.7, Nav1.8, Nav1.9), inflammasome complexes (NLRP3), the kynurenine pathway, prostaglandins, and bradykinin-mediated signaling. Emerging therapeutics such as selective Nav channel blockers, NLRP3 inhibitors, kynurenine pathway modulators, EP receptor antagonists, and bradykinin receptor antagonists offer promising alternatives to opioids. Despite challenges in clinical translation, these developments signal a paradigm shift in pain management, with precision-focused therapies poised to address unmet needs. This review emphasizes the importance of integrating molecular insights into the development of safer, more effective analgesics, setting the stage for transformative advancements in non-opioid pain relief.
Introduction
Pain, as defined by the International Association for the Study of Pain (IASP), is an “unpleasant sensory and emotional experience associated with actual or potential tissue damage.” 1 It is a highly individualized and complex phenomenon, shaped by genetic, environmental, and psychological factors. Pain can manifest in various forms – acute or chronic, nociceptive or neuropathic, inflammatory or non-inflammatory – each with distinct mechanisms and implications. 2 Its biological role is primarily protective, as seen in nociceptive pain, where tissue damage activates sensory pathways to prevent further harm.3,4 In contrast, neuropathic pain, often resistant to conventional analgesics, arises from nerve damage and lacks this protective function, posing challenges in management.5–7
Traditional pain management has relied heavily on opioids due to their effectiveness in both acute and chronic pain cases. However, the opioid epidemic, particularly pronounced in the United States, has revealed the severe risks associated with opioids, including addiction, tolerance, and overdose.8–11 The growing prevalence of prescription misuse and synthetic opioids, such as fentanyl, has led to a critical reassessment of pain management practices, with a focus on developing safer, non-opioid alternatives. 5
This review explores emerging approaches targeting molecular mechanisms of pain with the goal of reducing opioid reliance. While our focus is on molecular signaling pathways involved in pain, specifically the pathways that influence one of inflammation’s cardinal signs – dolor (pain) – it is not intended to be exhaustive. Instead, we highlight promising non-opioid targets such as voltage-gated sodium channels, inflammasomes, the kynurenine pathway, prostaglandins, and bradykinin, which are currently under investigation.3,12–20 These pathways represent potential new directions in pain pharmacology, addressing both the challenges of chronic pain and the complexities inherent in pain modulation.
A brief overview of pain mechanisms
Pain can be broadly categorized into nociceptive, neuropathic, and inflammatory types, each with distinct biochemical pathways and implications for treatment. Nociceptive pain, typically acute, is triggered by tissue damage and mediated by specialized sensory fibers that transmit pain signals to the CNS. Aδ fibers, for example, are myelinated and rapidly respond to thermal and mechanical stimuli, producing sharp, localized pain, while unmyelinated C fibers are slower, conveying dull, aching sensations often associated with prolonged inflammation or neuropathic conditions.21,22 Neuropathic pain, on the other hand, results from damage to the somatosensory nervous system, which can impact the CNS or peripheral nerves. 23 It is often associated with conditions like diabetes or trauma and is characterized by symptoms such as allodynia, where normally non-painful stimuli cause pain. Ion channel dysregulation, particularly involving Sodium Gated Voltage Channels (SGVC, Nav) in the dorsal root ganglion (DRG), contributes to excessive nerve firing and heightened sensitivity in neuropathic pain. 13
Inflammatory pain, a critical component of the body’s response to injury, serves to alert and mobilize the immune system to begin the healing process. This type of pain is mediated by a cascade of chemical mediators like prostaglandins, kinins, and cytokines, which amplify nociceptive signaling.15,16 Prolonged inflammation may lead to central sensitization, creating a state of hyper-responsiveness to pain. 14 Recognizing the interplay among these pain types and pathways will be essential for developing advanced therapies that can manage chronic pain effectively without compromising the body’s innate warning system.
In summary, this review will focus on non-opioid targets within these pathways, with particular emphasis on the mechanisms involved in inflammatory pain signaling. Although inflammation and pain are intricately connected, the review will not be limited to inflammatory pain alone; rather, it will encompass various signaling pathways relevant to both acute and chronic pain. Through these explorations, we aim to illuminate promising avenues for future pain relief strategies that address both the complexity of pain and the limitations of current treatment options.
Voltage-gated sodium (Nav) channels in pain mechanisms
Voltage-gated sodium channels are essential for initiating and propagating action potentials in excitable cells, including neurons and certain immune cells. These channels, numbered Nav1.1 through Nav1.9, respond to changes in membrane potential by briefly opening to allow sodium ions to flow into the cell, depolarizing the cell membrane and triggering action potentials that relay signals throughout the nervous system. This process is crucial in pain signaling, where Nav channels modulate the excitability of pain-sensing neurons.13,24
Structurally, Nav channels consist of four domains (DI–DIV), each with six transmembrane segments (S1–S6). The S4 segments act as voltage sensors, detecting changes in membrane potential to open the channel, while the S5 and S6 segments form a pore that selectively permits sodium ions to pass through.25,26 See Catterall 26 for more detail on the sodium channel structure. This architecture is central to the generation and conduction of action potentials, making these channels vital targets in pain pathways, particularly within neurons involved in nociceptive and inflammatory pain signaling.
Among the Nav subtypes, Nav1.7, Nav1.8, and Nav1.9 are notably linked to pain modulation.26,27 Nav1.7, encoded by the SCN9A gene, has emerged as a key player in pain perception, with mutations causing either congenital insensitivity to pain or heightened pain disorders like erythromelalgia.28,29 Inhibitors of Nav1.7 target the S6 region to stabilize the channel in an inactive state, reducing neuronal excitability and pain transmission.27,28 Similarly, Nav1.8 and Nav1.9 are implicated in chronic and inflammatory pain.26,27 Nav1.8, primarily expressed in sensory neurons, mediates responses to painful stimuli, while Nav1.9 plays a role in sensitization to chronic inflammation. 30 Together, these channels have slower inactivation profiles, making them promising targets for new therapies aimed at managing persistent pain.
Additionally, Nav channel function can be modulated through intracellular post translational modifications such as phosphorylation, affecting their activity and localization within the cell. 31 This regulatory mechanism is an emerging focus in research to develop selective sodium channel inhibitors for pain relief, offering potential alternatives to opioid-based treatments. 31
Nav channels as therapeutic targets in pain management
Recent advancements in Nav channel blockers highlight their potential as non-addictive options for pain management. 27 In particular, Nav1.7 has garnered attention due to its role in pain transmission. Inhibitors like VX-150 (Vertex), selectively target Nav1.7, demonstrating significant efficacy in clinical studies for chronic pain conditions such as osteoarthritis, small fiber neuropathy, and acute pain in post-surgical settings. 32 Another Nav1.7-targeting compound, Vixotrigine (Convergence), has been under clinical evaluation for a range of pain conditions, including trigeminal neuralgia, lumbosacral radiculopathy, and diabetic neuropathy. 27 Early findings suggest it may provide effective pain relief without the risks associated with opioids.33,34
Beyond Nav1.7, Nav1.8, and Nav1.9 inhibitors are also advancing as potential therapies for chronic and inflammatory pain. For example, Suzetrigine, previously known as VX-548 (Vertex) targets Nav1.8 and has received FDA priority review/approval for moderate-to-severe acute pain, potentially introducing the first new class of acute pain medications in decades.32,35,36
Another novel agent, ANP-230, previously DSP-2230 (Sumitomo Pharma), characterized by Kamei et al., 32 targets Nav1.7, Nav1.8, and Nav1.9 and is in clinical development for peripheral neuropathic pain. This compound’s unique profile differentiates it from existing therapies and may enhance its effectiveness in treating persistent pain conditions. 32
As ongoing research deepens our understanding of Nav channel mechanisms, these targeted inhibitors hold promise for non-opioid pain relief. By addressing the pain pathways modulated by Nav1.7, Nav1.8, and Nav1.9, these agents provide hope for managing chronic pain with reduced risk of addiction, marking a significant shift in contemporary pain management strategies.
The NLRP3 inflammasome complex and pain modulation
The NLRP3 inflammasome is a critical intracellular multiprotein complex that responds to cellular danger signals, playing a central role in immune responses and pain signaling (Figure 1). Upon activation, it triggers caspase-1, leading to the release of pro-inflammatory cytokines, notably IL-1β and IL-18, which contribute to the sensitization of nociceptors and amplification of pain signals. 17 Pattern recognition receptors (PRRs), specifically NOD-like receptors, detect both pathogen-associated and damage-associated molecular patterns that initiate inflammasome assembly. 17 This complex has been implicated in various pain-related disorders, including multiple sclerosis, rheumatoid arthritis and others, where chronic inflammation exacerbates pain.17,18,37 Given its role in both neuroinflammation and peripheral inflammation, the NLRP3 inflammasome presents a promising therapeutic target for managing chronic pain while avoiding opioid-related side effects. Below, we review the emerging clinical approaches targeting NLRP3 and its associated pathways in pain management.

The inflammasome pathway in pain modulation.
Drugs targeting the inflammasome
The NLRP3 inflammasome has become a focal point for developing non-opioid therapies for chronic inflammatory pain. Several inhibitors, each with distinct mechanisms, are being tested for their effectiveness in reducing inflammation and modulating pain.
One promising compound, NT-0249 (NodThera) is a novel NLRP3 inhibitor that has shown robust specificity for NLRP3-related inflammation. Pharmacological studies, such as the work by Doedens et al., 38 demonstrate that NT-0249 dose-dependently decreases IL-1β levels in tissue samples, signaling its potential for treating chronic inflammatory pain.
Another NLRP3 inhibitor, MCC950, also known as CP-456,773, (Pfizer) has shown efficacy in preclinical models, especially for preventing the transition from acute to chronic pain, as evidenced in hyperalgesia priming models.17,39–42 In addition, this compound has shown meaningful outcomes in IL-1B-treated chondrocytes, specifically by suppressing NLRP3 and cleaved caspase-1. 37 Despite its promise, MCC950 faced challenges in clinical trials due to safety concerns, including drug-induced liver injury during a Phase II trial for rheumatoid arthritis. 37 Recent derivatives of this structure: SZYIL1 (Zydus formerly known as Cadila Healthcare), selnoflast (Roche), and emlenoflast (Roche/Inflazome) are all currently being studied in various clinical trials. 37 These findings highlight the importance of evaluating both the efficacy and safety of inflammasome inhibitors in clinical development.
Alternative approaches targeting inflammatory pathways
While NLRP3 inhibitors like NT-0249 and MCC950 show potential, their safety challenges have prompted exploration of alternative targets within the inflammasome pathway. One such target is the P2X7 Receptor (P2X7R) a purine receptor, a key player in pain and inflammation.
The P2X7 receptor responds to high levels of extracellular ATP, typically released from damaged cells, to facilitate ion influx and inflammasome activation. This signaling pathway enhances the production of pro-inflammatory cytokines like IL-1β and IL-18, which both drive pain and inflammatory responses. 43 AZD9056 (AstraZeneca), a selective P2X7R antagonist has demonstrated effectiveness in reducing inflammatory pain by modulating nociceptive transmission. 44 Initial clinical trials indicated AZD9056’s potential as a non-opioid analgesic, though studies were terminated in phase II trials due to lack of efficacy.45,46
Additional targets in inflammasome-related pain management
Expanding beyond direct inflammasome inhibitors, research has also focused on other molecular targets associated with NLRP3 activation and downstream pain signaling pathways.
Cathepsin B is a lysosomal protease critical for inflammasome activation, as it facilitates NLRP3 assembly through lysosomal damage, amplifying inflammatory responses. Cathepsin B also contributes to neuroinflammation, linking it to pain conditions through extracellular matrix breakdown and immune modulation. 47
Cathepsin B inhibitors have recently gained attention as potential pain modulators in the context of inflammatory diseases. One such inhibitor, CA-074Me (not in clinical trials, manufactured for research by multiple companies Millipore-sigma, Selleck chemicals, APExBIO, BioCrick, AbMole Biosciences) selectively targets cathepsin B and has demonstrated promise in preclinical models. Studies show that CA-074Me effectively reduces hypersensitivity and inflammation in animal models by inhibiting the proteolytic activity of cathepsin B, which is linked to inflammatory pain pathways.17,47,48 Despite its efficacy in preclinical studies, there is limited information on CA-074Me advancing into later-stage clinical trials for pain management. Additionally, Aloxistatin, formerly known as E64d (Taisho Pharmaceuticals), a broad-spectrum cathepsin inhibitor, has been explored in preclinical models for various conditions, including inflammation and traumatic brain injury. While some studies have indicated its potential in reducing inflammatory pain by inhibiting cathepsin B activity, E64d has not progressed to clinical trials for pain management. 37 Its development has mainly been focused on other conditions, such as muscular dystrophy, and as of now, no pharmaceutical companies are actively investigating E64d for pain-related treatments. Further research would be required to explore its therapeutic viability for pain management.
Gasdermin D (GSDMD) plays a critical role in pyroptosis, a form of inflammatory cell death that significantly contributes to pain and inflammation. Upon activation by caspase-1 or caspase-5, GSDMD forms pores in the cell membrane, leading to cell lysis and the release of pro-inflammatory cytokines such as IL-1β and IL-18. This cascade amplifies pain responses, especially in conditions characterized by chronic inflammation, such as rheumatoid arthritis, osteoarthritis, and other inflammatory diseases. 37 Given its central role in inflammation, GSDMD has emerged as an important target for therapeutic intervention in pain management. Inhibiting GSDMD is considered a promising strategy to reduce inflammation and alleviate associated pain. Several compounds are currently being explored in preclinical studies as potential GSDMD inhibitors, including LDC7559, VX-765, and disulfiram. 49
LDC7559 (Max-Plank-Gesellschaft) is a small molecule designed to block GSDMD activation. This drug is still in the early stages of preclinical development, with a focus on modulating inflammatory pathways such as pyroptosis. While there is limited public information on the precise progression of LDC7559 through clinical trials, it is expected to play a significant role in treating conditions such as neuropathic pain and other inflammatory diseases. Preclinical research has shown that LDC7559 holds promise in modulating the pyroptotic pathway, which may reduce inflammation and alleviate symptoms of neuroinflammation. 43 However, LDC7559 has not yet entered human clinical trials, and further research is needed to evaluate its efficacy and safety in pain management.
VX-765/belnacasan (Vertex), is another promising drug under investigation for its ability to inhibit GSDMD activity and, consequently, reduce inflammation. VX-765 is a prodrug of VRT-043198, which inhibits caspase-1, preventing the activation of GSDMD and the subsequent release of inflammatory cytokines. 50 This drug has been evaluated in clinical trials for chronic inflammatory diseases and pain, with Phase 2 trials already underway for conditions such as epilepsy and other inflammatory disorders. 27 The application of VX-765 for pain management, particularly in diseases like rheumatoid arthritis and other inflammatory conditions, remains under investigation.
Disulfiram (Cantex), has been used for the treatment of alcohol use disorder under the brand name Antabuse, has been repurposed for its ability to block GSDMD activation. While disulfiram is not a direct GSDMD inhibitor, research has shown that it can inhibit the pore-forming ability of GSDMD, potentially reducing inflammation in diseases like rheumatoid arthritis and inflammatory bowel disease.44,49 Disulfiram’s repurposing for inflammation and pain management is being studied by multiple research groups and academic institutions. Although it is not currently being developed by a single company for exclusive use in pain management, ongoing clinical trials are exploring its potential to reduce inflammation and alleviate pain. Early preclinical and clinical studies suggest that disulfiram could become a valuable therapeutic option for managing chronic inflammation and pain, but more research is needed to fully understand its role in these conditions. 44
Future directions in non-opioid pain management
The exploration of NLRP3 inflammasome inhibitors, alongside P2X7R antagonists and regulators of Cathepsin B and Gasdermin D, highlights a paradigm shift in pain management. As clinical trials progress, these targeted therapies may provide effective alternatives to opioids by addressing the underlying inflammatory mechanisms of chronic pain. Understanding the complex roles of these molecular pathways in pain propagation could pave the way for safer, targeted treatments, reducing the reliance on opioid-based analgesics while enhancing the quality of life for patients suffering from chronic pain.
Kynurenine pathway in pain modulation
The kynurenine pathway, a major metabolic route for tryptophan, plays a critical role in pain modulation and neuroprotection (Figure 2). One of its key metabolites, kynurenic acid, acts as a non-competitive antagonist of NMDA receptors, thereby inhibiting glutamate-induced excitatory signaling. This inhibitory effect is particularly relevant in chronic pain, where NMDA receptor hyperactivity contributes to excitotoxic damage and heightened pain perception. By counteracting this excitotoxicity, kynurenic acid offers both neuroprotection and pain modulation, showing promise as a therapeutic target for neuroinflammatory diseases and chronic pain conditions.12,51
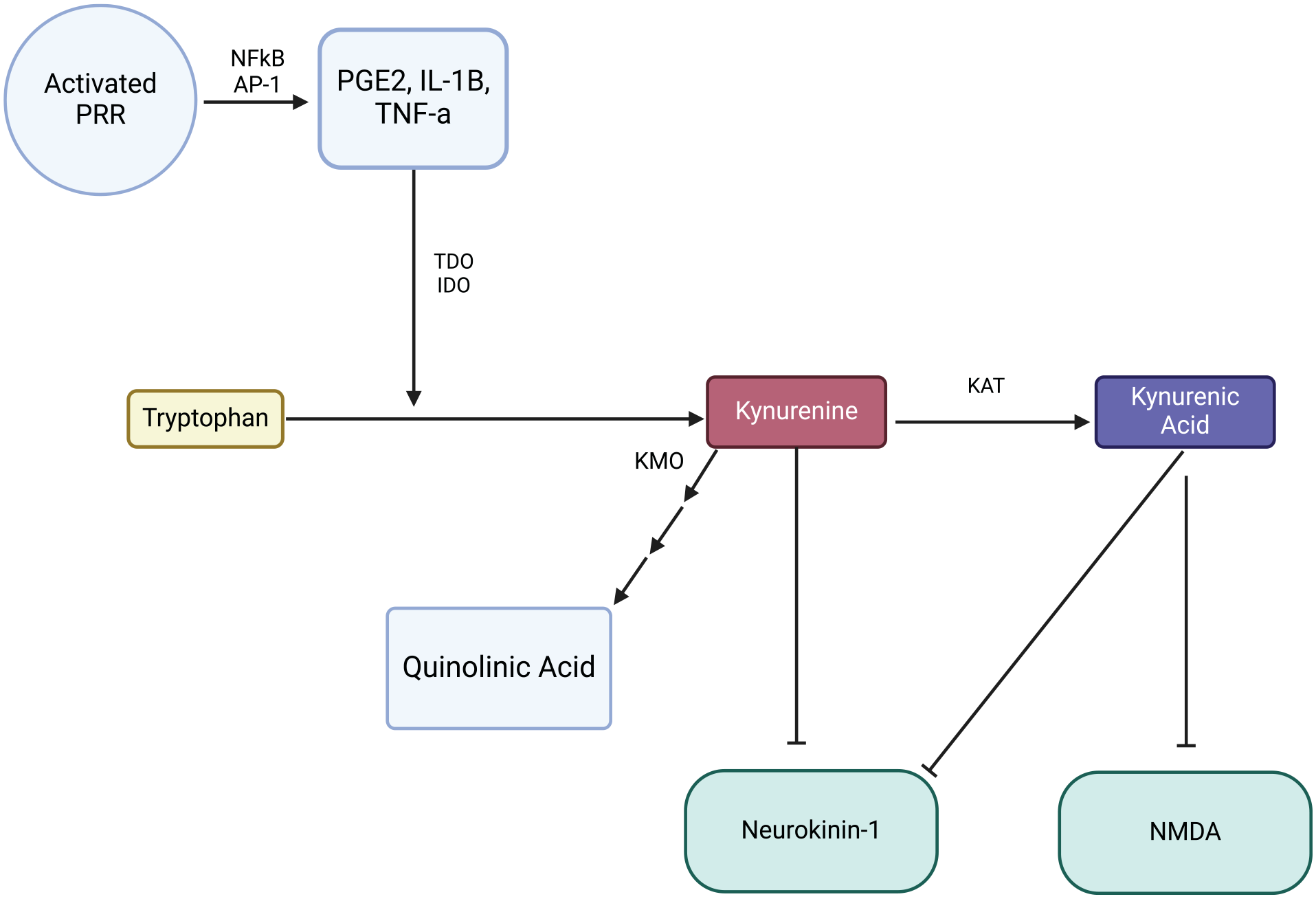
Mechanism of the kynurenine pathway in pain modulation.
Further, enhancing kynurenic acid levels via Kynurenine 3-monooxygenase (KMO) inhibitors has emerged as a non-opioid strategy to modulate NMDA receptor activity and reduce pain perception. 53 KMO inhibitors shift the kynurenine pathway toward the production of kynurenic acid rather than neurotoxic metabolites like quinolinic acid, providing an additional layer of neuroprotection. 53 Dysregulation in this pathway has been implicated in neuroinflammatory diseases, such as multiple sclerosis, where increased kynurenine metabolism exacerbates pain and neurological dysfunction.54,55
Kynurenine-3-monooxygenase (KMO) inhibitors in pain management
Among various targets in the kynurenine pathway, KMO has emerged as a promising therapeutic focus for managing neuropathic pain. KMO inhibitors, such as Ro 61-8048 (Merck KGaA) enhance kynurenic acid production while reducing the synthesis of neurotoxic quinolinic acid. This redirection of the pathway is crucial, as kynurenic acid is neuroprotective and antagonizes NMDA receptor activity, both mechanisms implicated in chronic pain and excitotoxicity. Kynurenic acid’s protective role in preventing neurodegeneration and its effects on pain modulation have been well-documented in both preclinical models and human studies. 55
A detailed study by Gao et al. 56 characterized Ro 61-8048 as a potent, selective KMO inhibitor with unique allosteric properties. The study described how Ro 61-8048, interacts with KMO to alter the conformation of the enzyme’s catalytic residue, thereby hindering substrate access and establishing its non-competitive inhibition. This selectivity allows for optimized therapeutic applications, making Ro 61-8048 a promising candidate in pain management. 56 Ro 61-8048’s versatility, demonstrated in models of neurodegenerative disorders, further highlights its potential applicability in pain modulation.
Other KMO inhibitors in development
Other KMO inhibitors currently under development include Ro 61-6048 (Merck KGaA) and JM6 (Merck KGaA). These compounds offer unique mechanisms to influence the kynurenine pathway and have demonstrated effectiveness in reducing neurogenic pain and improving cognitive function in animal models, including rodents. 12
Beyond KMO, inhibitors targeting other enzymes in the pathway, such as 1-D-Methyltryptophan or 1M-d-T (Lumos Pharma) an IDO-2 inhibitor and PCC0208009 (Bristol-Myers Squibb), an IDO-1 inhibitor, have shown potential for mitigating pain hypersensitivity. For instance, PCC0208009 has demonstrated the ability to alleviate pain and cognitive impairment in rats with spinal nerve ligation, indicating its dual therapeutic potential in pain and neuroprotection. 12
IDO-1 and IDO-2 inhibitors
The investigation of IDO-1 inhibitors like epacadostat, also named INCB024360 (Incyte Corporation), BMS-986205 (Bristol-Myers Squibb), and indoximod or NLG-8189 (Lumos Pharma, NewLink Genetics) continues, with studies showing varying degrees of efficacy in pain modulation and neuroinflammation models. 56 These inhibitors show promise for treating neuropathic pain and neuroinflammation, although clinical results have been mixed, particularly for epacadostat, which was halted in Phase 3 clinical trials for melanoma due to a lack of efficacy. Whereas indoximod has shown promising preliminary evidence of efficacy for phase II trials in the pediatric population undergoing chemotherapy. 57
Safety profiles and future directions
Notably, safety profiles for KMO and IDO inhibitors have been favorable in preclinical studies, with minimal adverse effects observed at therapeutic doses. This suggests a promising therapeutic index, making these compounds attractive candidates for future clinical trials.55,58 As the research on these compounds progresses, their potential for non-opioid treatments in chronic pain management continues to grow.
The ongoing development of KMO and IDO inhibitors highlight the kynurenine pathway’s viability as a target for neuropathic pain management. With a growing understanding of how these metabolic shifts impact pain and neuroinflammation, novel inhibitors like Ro 61-8048 and PCC0208009 offer promising, non-opioid approaches to treating chronic pain. The exploration of these pathways holds potential for more effective and safer treatments, advancing pain management beyond traditional therapies and toward precision medicine.
Prostaglandins and pain
Prostaglandins, particularly Prostaglandin E2 (PGE2), play a central role in the pain and inflammation response (Figure 3). Synthesized from arachidonic acid via the enzymes phospholipase A2 (PLA2) and cyclooxygenase (COX), PGE2 contributes to pain by sensitizing nociceptors, the pain-sensing neurons, thereby increasing pain sensitivity in inflamed tissues. 59 In inflammatory conditions, elevated levels of PGE2 amplify pain signaling, making it a significant target for both acute and chronic pain therapies.

The role of PGE2 and EP receptor signaling in pain modulation via Nav1.7 channels.
Targeting PGE2 and EP receptor antagonists for pain management
As a critical mediator of inflammation and pain, PGE2 has traditionally been targeted through non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit COX enzymes to reduce prostaglandin synthesis. While effective, NSAIDs can have widespread effects, often resulting in gastrointestinal, cardiovascular, and renal side effects due to the inhibition of all prostaglandin synthesis. 60 Selective targeting of PGE2 receptors – the EP receptors – presents a more focused approach with potentially fewer side effects.
EP2 receptor antagonists
The EP2 receptor is one of the main receptors implicated in pain signaling. PF-04418948 (Pfizer), an EP2-selective antagonist, was initially investigated for its potential to manage pain with fewer side effects than NSAIDs. 61 However, clinical development of PF-04418948 was discontinued due to safety and efficacy concerns that did not meet the requirements for progressing in trials. 61
EP4 receptor antagonists
Among the EP receptors, the EP4 receptor has gained particular attention as a promising target. Grapiprant known also as CJ-023,423 (Arrys Therapeutics) an EP4 receptor antagonist, selectively inhibits PGE2 signaling at this receptor, thereby reducing pain and inflammation while avoiding the broad suppression of COX enzymes. Currently, Grapiprant is in trials for treating osteoarthritis-related pain in veterinary medicine, where it has shown promise in reducing pain and improving function with a favorable safety profile approved by the FDA.62,63
Other EP4 antagonists, such as ONO-4232, (ONO Pharmaceutical) are under development and preclinical investigation for their potential in pain relief, targeting inflammatory and nociceptive pathways specifically mediated by EP4 receptor activation. 16 This drug, along with Grapiprant, exemplify the focus on more selective interventions within the prostaglandin pathway, addressing pain without the systemic impact associated with NSAIDs.
Future directions in PGE2 and prostaglandin-related pain therapies
The exploration of PGE2 and its receptors, particularly EP2 and EP4, highlights the ongoing shift toward precision pain management. By selectively targeting PGE2 receptors, researchers aim to modulate pain pathways directly related to inflammation, providing effective pain relief with a more favorable safety profile. These selective approaches offer hope for chronic pain sufferers, particularly those with inflammatory conditions such as osteoarthritis, who may benefit from tailored therapies that reduce pain and inflammation while minimizing systemic side effects. As the development of receptor-specific drugs continues, PGE2-targeted therapies are poised to play an increasingly prominent role in non-opioid pain management.
Bradykinin and pain
Bradykinin, a peptide generated during inflammation, is a critical mediator of pain that acts through B1 and B2 receptors on sensory neurons (Figure 4). Upon tissue injury, bradykinin is released and binds to these receptors, amplifying nociceptive signals and contributing to both acute and chronic inflammatory pain. 64 Bradykinin plays a dual role in modulating inflammatory and neuropathic pain, as it sensitizes pain pathways and enhances inflammatory responses, making it a significant target for non-opioid analgesic strategies.
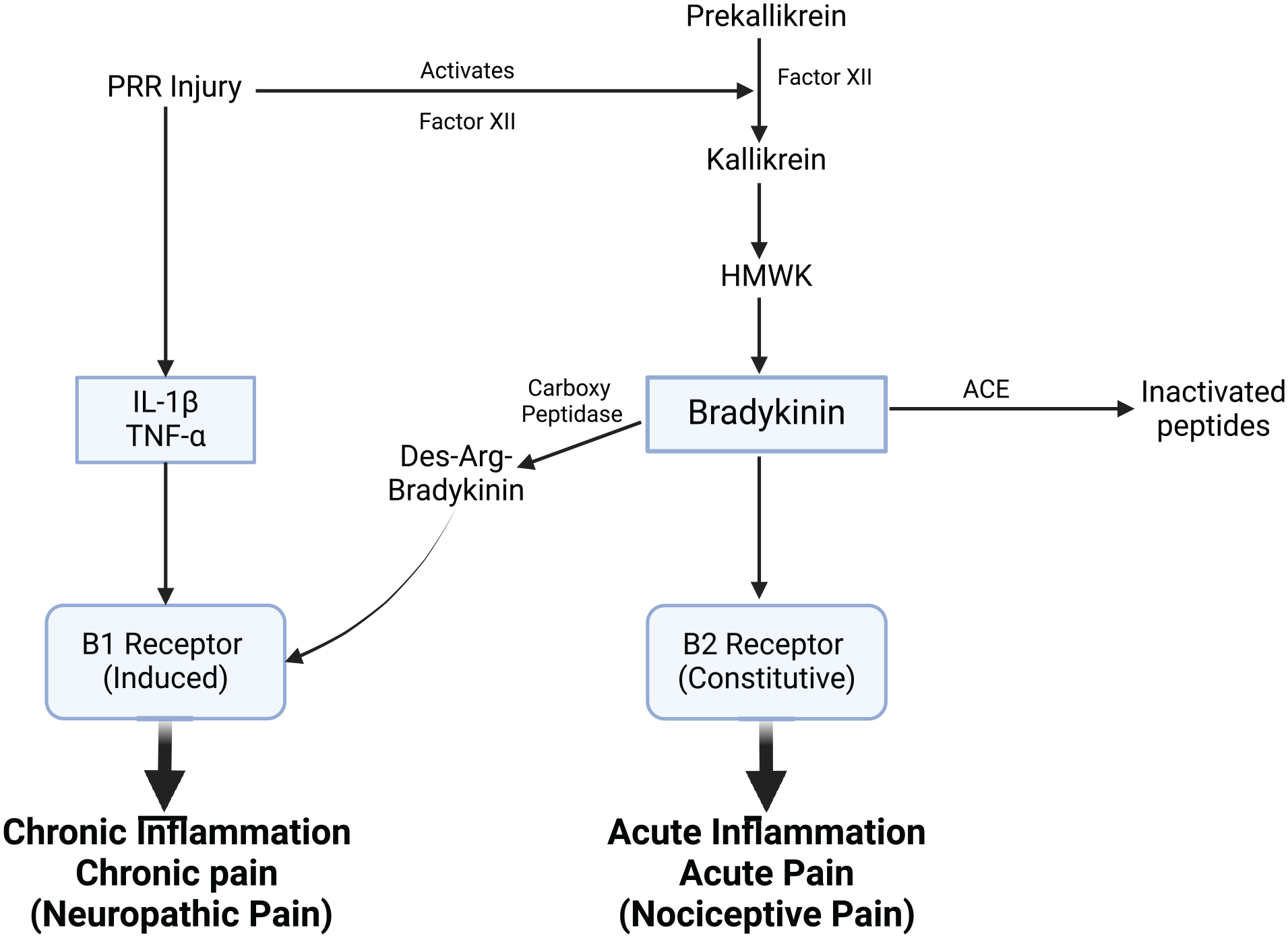
Bradykinin pathway and receptor signaling in inflammatory pain modulation.
The activation of B1 receptors initiates downstream signaling cascades involving mitogen-activated protein kinases (MAPKs) and nitric oxide (NO) production through nitric oxide synthase (NOS), enhancing inflammatory responses and sensitizing pain pathways. In contrast, binding to B2 receptors activates phospholipase C (PLC), which leads to the production of inositol trisphosphate (IP3) and diacylglycerol (DAG). This in turn raises intracellular calcium (Ca²⁺) levels, activating protein kinase C (PKC), and further amplifying pain signaling. Additionally, angiotensin-converting enzyme (ACE) degrades bradykinin into inactive peptides, providing a regulatory mechanism to modulate its effects. The net effect of bradykinin signaling through these pathways is the promotion of pain sensitization and sustained inflammatory responses (Figure 4).
Bradykinin receptor antagonists in pain management
Targeting bradykinin receptors, particularly B1 and B2, presents a promising strategy for managing pain without opioids.
The B2 receptor, which is constitutively expressed, plays a central role in acute pain through its release of pro-inflammatory mediators. This receptor is highly active in nociceptive pain conditions such as arthritis and post-surgical pain. 65 Icatibant (Takeda Pharmaceuticals), a B2 receptor antagonist initially developed for hereditary angioedema, is being evaluated in clinical trials for its potential in treating acute inflammatory pain. 66 Icatibant works by blocking B2 receptor activity, effectively reducing the inflammatory signaling that underlies acute pain. 66
Unlike B2, the B1 receptor is upregulated in response to prolonged inflammation and chronic pain states. Its activation is associated with hyperalgesia and allodynia, common in chronic pain conditions like rheumatoid arthritis and fibromyalgia. 64 Recent clinical trials have shown that B1 receptor antagonists can significantly reduce inflammatory pain by specifically targeting these upregulated receptors, positioning them as promising candidates for non-opioid pain therapies. 64
Investigational B1 antagonists are currently in early-phase trials for managing chronic inflammatory conditions, demonstrating potential in reducing pain and improving quality of life for patients with persistent pain. 67 There have been multiple compounds that have entered clinical trial, many only to be terminated by the manufacturers with no superiority to placebo. However, B1R antagonist BAY 2395840 (Bayer) recently was studied in a phase 2a study to evaluate efficacy and safety in diabetic patients with neuropathic pain. 67
Both B1 and B2 receptors adapt to sustained inflammatory stimuli, which contributes to the chronic nature of pain by maintaining heightened pain sensitivity. This receptor adaptation reinforces the persistence of pain, suggesting that modulating bradykinin receptor signaling may restore normal pain perception and provide relief from hyperalgesia.19,68
Saline in pain modulation
Interestingly, saline solutions have been shown to influence pain signaling pathways, providing symptomatic relief in various pain contexts. Studies have demonstrated that hypertonic saline can exacerbate nociceptive pain by stimulating nociceptors, whereas hypotonic saline may alleviate neuropathic pain. This effect of saline solutions reinforce their potential in modulating pain during clinical interventions, such as fluid resuscitation, and highlights the nuanced role of electrolyte balance in pain management. 69 Further research may explore saline’s role in pain modulation, particularly in combination with other non-opioid treatments.
Key interactions among pain modulation pathways: implications for drug development
Pain is orchestrated through an intricate network of signaling pathways, many of which are interdependent and exhibit extensive crosstalk. Understanding these interactions is crucial for the development of effective non-opioid analgesics, as targeting isolated components can lead to suboptimal therapeutic outcomes. This section outlines key interactions among the pathways reviewed, emphasizing their impact on drug efficacy and the translational challenges faced in clinical development.
Prostaglandins and voltage-gated sodium channels (Navs)
Prostaglandins, particularly PGE2, not only mediate inflammation but also modulate neuronal excitability. PGE2 sensitizes nociceptors by lowering the activation threshold of Nav channels such as Nav1.7, Nav1.8, and Nav1.9 through EP receptor signaling. This interaction amplifies pain signaling in inflammatory conditions. Consequently, targeting Nav channels alone may not fully alleviate pain if prostaglandin-mediated sensitization remains unchecked, highlighting the potential benefit of combination therapies targeting both pathways.
Inflammasomes (NLRP3) and the kynurenine pathway
Activation of the NLRP3 inflammasome leads to the release of pro-inflammatory cytokines like IL-1β, which can influence the kynurenine pathway by upregulating indoleamine 2,3-dioxygenase (IDO). This enzyme shifts tryptophan metabolism toward neuroactive kynurenine metabolites, some of which (e.g. quinolinic acid) are neurotoxic and can exacerbate neuropathic pain. Thus, inflammasome inhibitors may have downstream effects on kynurenine metabolism, offering a broader anti-nociceptive impact than initially anticipated.
Bradykinin signaling and prostaglandin synthesis
Bradykinin, a potent pain mediator, stimulates phospholipase A2 activity, increasing arachidonic acid release and subsequent prostaglandin synthesis via COX enzymes. This cascade not only promotes inflammation but also potentiates bradykinin receptor sensitivity, creating a feedback loop that sustains pain. Targeting bradykinin receptors can, therefore, indirectly reduce prostaglandin production, suggesting a synergistic potential when combined with COX inhibitors or EP receptor antagonists.
Sodium channels and inflammasome activation
Recent evidence suggests that sodium influx through Nav channels can modulate inflammasome activity. For instance, altered ionic homeostasis due to Nav1.7 dysfunction may influence NLRP3 activation thresholds, linking neuronal excitability with innate immune responses. This interaction underscores the dual role of Nav channels in both neuronal and inflammatory processes, complicating the development of selective inhibitors that avoid unintended immunomodulatory effects.
Kynurenine pathway and neuroinflammation
Neuroinflammation, driven by glial activation and cytokine release, can upregulate the kynurenine pathway, creating a vicious cycle where inflammatory signals enhance the production of neurotoxic metabolites. This feedback loop contributes to chronic pain states, particularly in neuropathic conditions. Modulators of the kynurenine pathway may thus exert their analgesic effects not only by altering metabolite levels but also by dampening neuroinflammatory responses.
Implications for drug development
The extensive crosstalk among these pathways highlights the limitations of monotherapies targeting single molecules. Compensatory mechanisms often undermine the efficacy of highly selective drugs, contributing to high attrition rates in clinical trials. A systems biology approach, incorporating multi-target strategies and combination therapies, may offer a more robust solution. Additionally, identifying biomarkers that reflect pathway interactions could improve patient stratification and therapeutic outcomes.
Conclusions
The urgent need for non-opioid pain therapies is apparent considering the ongoing opioid epidemic. By targeting key molecular pathways – sodium channels, inflammasomes, the kynurenine pathway, prostaglandins, and bradykinin – researchers are pioneering new classes of analgesics that offer effective pain relief with reduced risk of addiction and adverse effects. Clinical trials for drugs targeting these pathways are underway, with early results suggesting these therapies may significantly shift the landscape of pain management, providing safer and more sustainable options for patients.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.