
Abstract
Fabry disease (FD) is a X-linked lysosomal storage disorder caused by deficient function of the alpha-galactosidase A (α-GalA) enzyme. α-GalA deficiency leads to multisystemic clinical manifestations caused by the preferential accumulation of globotriaosylceramide (Gb3). A hallmark symptom of FD patients is neuropathic pain that appears in the early stage of the disease as a result of peripheral small fiber damage. Previous studies have shown that Acetyl-L-carnitine (ALC) has neuroprotective, neurotrophic, and analgesic activity in animal models of neuropathic pain. To study the action of ALC on neuropathic pain associated with FD, we treated α-GalA gene null mice (α-GalA(-/0)) with ALC for 30 days. In α-Gal KO mice, ALC treatment induced acute and long-lasting analgesia, which persisted 1 month after drug withdrawal. This effect was antagonized by single administration of LY341495, an orthosteric antagonist of mGlu2/3 metabotropic glutamate receptors. We also found an up-regulation of mGlu2 receptors in cultured DRG neurons isolated from 30-day ALC-treated α-GalA KO mice. However, the up-regulation of mGlu2 receptors was no longer present in DRG neurons isolated 30 days after the end of treatment. Taken together, these findings suggest that ALC induces analgesia in an animal model of FD by up-regulating mGlu2 receptors, and that analgesia is maintained by additional mechanisms after ALC withdrawal. ALC might represent a valuable pharmacological strategy to reduce pain in FD patients.
Introduction
Neuropathic pain is induced by morphological and/or functional abnormalities of the somatosensory system, and develops as a result of a cascade of biochemical events and adaptive mechanisms, which cause dysregulation of sensory neurons and maladaptive plasticity within the nociceptive system. Chronic pain is sustained by a malfunction of the pain neuraxis, which alters the processing of nociceptive signaling in such a way that pain is felt even in the absence of detectable noxious or inflammatory inputs, and responses to innocuous and noxious stimuli are enhanced. 1 These alterations are known as peripheral and central sensitization, which reflect an amplified response of peripheral nociceptors and an increased synaptic transmission in the pain pathways, respectively, resulting in a reduction in pain thresholds and an amplification of pain responses. Neuropathic pain is a hallmark of a wide group of peripheral neuropathies, including Anderson-Fabry disease (FD). FD is a multi-systemic X-linked lysosomal storage disorder (LSD), caused by a reduced or absent function of the enzyme α-galactosidase A (α-GalA), a lysosomal hydrolase responsible for glycosphingolipid metabolism.2,3 To date, over 1000 mutations of GLA gene with a possible linkage to FD have been identified. 4
Neuropathic pain is directly related to the loss of enzymatic activity and it is considered the earliest symptom of FD. Furthermore, it could be acute/episodic or chronic.5–7 Episodic pain includes allodynia (i.e., pain caused by a normally non-painful stimulus) and hyperalgesia (i.e., an exaggerated painful sensation in response to a noxious stimulus).8,9 Chronic pain associated with FD form appears as acroparesthesia (i.e., a burning or tingling sensation in both hands and feet). 10
Glutamate, the major excitatory neurotransmitter in the mammalian CNS, plays a key role in pain transmission and in nociceptive sensitization. Glutamate activates both ionotopic and metabotropic receptors (iGlu and mGlu receptors, respectively). Nowadays, eight mGlu receptor subtypes have been cloned and subdivided into three groups on the basis of their sequence similarity, pharmacological profile, and transduction mechanisms.11,12 Group I mGlu receptors (mGlu1 and-5 receptors) are coupled to Gq/11 proteins, and their activation likely contributes to the induction and expression of nociceptive sensitization as a result of intracellular Ca2+ mobilization and protein kinase C activation. In contrast, mGlu2 and mGlu4 receptors negatively modulate pain transmission by restraining glutamate release from presynaptic terminals in the dorsal horns of the spinal cord and, presumably, in other stations of the pain neuraxis.13–15 mGlu7 and mGlu8 receptors have also been involved in the regulation of pain under pathological conditions. 16 Accordingly, activation of mGlu8 receptors in the central nucleus of the amygdala relieves inflammatory pain, 17 whereas mGlu7 receptor blockade in the ventrolateral periaqueductal grey has antinociceptive effects in the formalin model of inflammatory pain and in the spare nerve injury model of neuropathic pain. 18 In addition, both mGlu7 and mGlu8 receptors in the dorsal striatum are involved in the modulation of pain thresholds.19–21
Acetyl-L-carnitine (ALC) is the major ester of L-carnitine (LC), a derivative of the amino acid lysine and methionine. ALC is naturally produced by ALC-transferase (CAT), an enzyme located in the mitochondrial matrix of a host of cellular types, and stimulates the catabolism of long fatty acids (β-oxidation) by facilitating the transport of fatty acids in mitochondria.22,23 ALC is known to induce neuroprotective, neurotrophic, and analgesic effects in experimental animal models of neuropathic pain, including the chronic constriction injury (CCI) model in mice, and, therefore represents a valuable therapeutic option for painful peripheral neuropathies, such as diabetic neuropathy.24–27 ALC-induced analgesia is mediated by epigenetic mechanisms based on acetylation of histones and transcription factors. By acetylating NFkB/p65 and H3 histone, ALC induces the expression of the GRM2 gene encoding the mGlu2 receptor in dorsal root ganglia and dorsal horns of the spinal cord.14,28–30 Histone deacetylase (HDAC) inhibitors can also cause analgesia by enhancing the expression of mGlu2 receptors in the spinal cord. 14 As a result of an epigenetic mechanism, ALC-induced analgesia is long-lasting and persists weeks after drug withdrawal in models of chronic inflammatory o neuropathic pain. 31
Based on these evidences, the first aim of our study was to ascertain whether ALC could induce a long-lasting analgesia in a mouse model of FD. The second goal of this study was to establish whether this long-lasting analgesia is correlated to an increased expression of mGlu2 receptors in the spinal cord.
Materials and methods
Animal model
Heterozygous female α-Gal A (+/−) and wild type α-Gal A (+/0) male mice (same JAX strain B6; 129Gla-tm1Kul/J) purchased from Charles River Laboratories Italia s.r.l. (Jackson Laboratory; Bar Harbor, ME, USA) were crossed to give the F1 generation.31,32 These mice were used from the F1 to the F4 generation as heterozygous females (+/−, B6; 129-Glatm1Kul/J) crossed with wild type males (+/0, B6; 129-Glatm1Kul/J) of the same genetic background (B6; 129-Glatm1Kul/J). From the F4 generation, we obtained homozygous females α-Gal A (−/−) and hemizygous males α-Gal A(-/0) mice that were compared to α-Gal (+/+ and +/0) as controls. The two homozygous/hemizygous knock-out and wild type groups were separated after at least 4 generations that is known to be enough to stabilize the background and all the experiments were performed after more than 10 generations. 33 Therefore, well-established homozygous α-Gal A (−/−) and hemizygous α-Gal A(-/0) mice compared to α-gal (+/+ and +/0) controls were used. Because the X inactivation process might have impaired the reproducibility of our model, and because FD is more severe in male patients,34,35 we decided to use exclusively male α-Gal A (-/0) mice in our study.
Mice were housed in groups of six in individually ventilated cages (Tecniplast, Italia) with water and food ad libitum in controlled environmental conditions: lights on from 7.00 a.m. to 7.00 p.m., 22 ± 2°C temperature and 65% humidity. Only α-GalA (+/0) (WT) and α-GalA (−/0) (KO) male mice were used. Behavioral experiments were carried out at the Department of Medical and Clinical Sciences (DIMEC), University of Bologna, with the approval of the local ethical committee (Veterinary Service of the University of Bologna) and in agreement with the National Animal Welfare Act. All efforts were made to minimize animal suffering and the number of animals used was kept to a minimum by the experimental design. All the procedures followed in this work complied with the European Community Council Directive of 24 November 1986 (86/609/EEC) and were approved by the Ethical committee of the University of Bologna (prot. N. 141/2019PR). The age of animals used for behavioral and immunohistochemistry experiments was in the range of two and three months of age.
Treatments
Timeline of behavioral and ex-vivo experiments.


Behavioral procedures
Behavioral tests were conducted between 09:00 am and 17:00 PM. Mice were habituated to the experimental room 1 h before the tests. Behavioral analysis was performed with a blind procedure.
Mechanical hyperalgesia
Mice were placed in test cages with a metal grid bottom at least 2 h prior to testing to allow accommodation in the novel environment. Paw withdrawal latency to mechanical stimulation was assessed with an automated testing device consisting of a steel rod (2 mm) that was pushed with electronic ascending force against the plantar surface of the hind paw with increasing force until the paw was withdrawn (Dynamic Plantar Aesthesiometer, Ugo Basile, Varese, Italy). A linear increase in force to 5 g was applied over 10 s after which the force remained constant. An interval of 120 sec was used during testing. When a mouse withdrew its hind paw, the mechanical stimulus was automatically withdrawn and the force recorded to the nearest 0.1 g. The paw withdrawal latency and actual force at the time of paw withdrawal reflex were calculated as the mean of 5 consecutive trials.
Hot plate test
Mice were positioned in the experimental room 1 h before the test. Each mouse was placed into a transparent Plexiglas beaker of 60 × 18 cm to avoid animals escaped from the plate, the temperature of which was set at 52 ± 0.1°C by using a thermo-regulated heated plate (Ugo Basile, Varese, Italy). The time (in seconds) between the placement of the animal and the first response: paw licking/fanning or jumping was measured as latency. A 30-s cut-off was used to prevent tissue damage. Measurements started 45 min after administration of LY 341495. Mice were investigated by observers who were blinded to animal treatment.
Preparation of rat dorsal root ganglion neuron
Primary cultures of DRG neurons were prepared from adult 8–12 weeks old males, according to previously described protocols with some modifications. 35 Mice were anesthetized by halothane prior to decapitation. All ganglia were removed from each mouse and transferred in ice cold DPBS 1x (Gibco) and the roots were cut using microdissecting scissors. After rinsing in DMEM (Gibco), the ganglia were placed in DMEM containing 5000 U/mL type IV collagenase (Worthington) for 45–75 min at 37°C, 5% CO2, washed twice with FBS-containing medium, and then gently mechanically dissociated with passages through 0.5 mm and 0.6 mm sterile needles. Cells were centrifuged for 10 min at low speed and then appropriately diluted in 1 mL of DMEM medium containing 10% FBS (Gibco), 50 ng/mL NGF (Gibco), and 1.5 μg/mL cytosine β-D-arabinofuranoside, (AraC, Sigma). For protein extraction, 60.000 cells/5 ml were plated onto Poly-Lysine pre-coated culture dishes. For immunocytochemistry, 15.000 cell/ml were plated onto 18 mm round glass coverslips, pre-coated with poly-L-lysine and Laminin. Cell cultures were maintained in an incubator at 37°C, with 5% CO2 for different periods of time (for electrophysiological experiments, cells were used within the first 4 days after plating (DIV); for WB analysis and immunocytochemistry cells at 4 or 6 DIV were used. Cells were maintained in DMEM, supplemented with 10% FBS in the presence of 50 ng/mL NGF, and 1.5 μg/mL cytosine β-D-arabinofuranoside, (AraC, Sigma) to reduce glial cell expression. Half volume of medium was changed every second day.
Immunofluorescence analysis
DRG neurons were isolated from WT and KO mice treated with ALC or saline. Cells were seeded on coverslips coated with poly-d-lysine; after 48 h, cells were fixed for 10 min with 4% formaldehyde at room temperature. After washing (2 times for 10 min each) in PBS, they were blocked with 5% BSA, 0.05% Triton X-100 in PBS for 1 hr at RT, and then incubated overnight at 4°C with the following primary antibodies: rabbit anti-mGlu2/3 (1:500, Millipore) or guinea pig anti-PGP9.5 (1:1000, Millipore), diluted in 1% BSA in PBS with 0.05% Triton X-100. On the following day, preparations were washed 3 × 10 min with PBS 1X and the following secondary antibodies were applied for 2 h at RT in the dark: Alexa 488 donkey anti-rabbit (1:400, Jackson ImmunoResearch) and Alexa 568 gut anti-guinea pig (1:400, Jackson ImmunoResearch) diluted in 1% BSA in PBS with 0.05% Triton X-100. After incubation, washing for 3 × 10 min with PBS was followed by mounting into medium containing DAPI on coverslip. Images were taken by a confocal microscope (Nikon C1).
Immunohistochemical analysis
Samples were isolated from WT and KO mice. All animals were deeply anesthetized and perfused transcardially with 4% paraformaldehyde (Sigma) in phosphate-buffered saline (PBS) (0.01 M, pH 7.4); the spinal cords were extracted and subjected to postfixation in 0.4% paraformaldehyde in PBS at 4°C overnight. We replaced the fixative solution with cryoprotective solution of 30% sucrose (Sigma). Cryostat sections (25 μm thick) were obtained from lumbar (L4-L5) spinal cord segment (corresponding to T13 vertebral spine) and collected on slides (Superfrost Plus, Thermo Scientific). After washing (2 times for 5 min each) in 0.01 M PBS, we performed the antigen retrieval at 80°C in 10 mM sodium citrate buffer. Sections were washed 3 times for 10 min in 10 mM PBS and endogenous peroxidases was blocked with 0.3% H2O2 in methanol. After 2 washes in 10 mM PBS for 10 min, the slides were blocked with 5% BSA, 0.1% Triton X-100, PBS 1X for 1 hr at RT, and then incubated overnight at 4°C with rabbit anti-mGlu2/3 (1:50, Millipore) in 1% BSA in 1X PBS with 0.1% Triton X-100. On the following day, preparations were washed 3x10 min with PBS 1X and the following secondary antibody was applied for 1.5 h at RT in the dark: goat anti rabbit-HRP (Santa Crutz) diluted in 1% BSA in 1X PBS with 0.1% Triton X-100. After three washings of 10 min with PBS 1X, the signal was revealed through the DAB Substrate Kit (Vector Laboratories) for 5 minutes and washed in H2O. Afterward, slides were mounted with glycerol and images were taken by microscope (AxioImager M1, (Carl Zeiss, Oberkochen, Germany) and analyzed (AxioCam MRc5, Carl Zeiss) software (AxioVision Rel 4.8, Carl Zeiss).
Statistical analysis
Statistical analysis.

Results
Effect of 30-day treatment with ALC on mechanical pain threshold in α-GalA (+/0) (WT) and α-GalA (−/0) (KO) mice
We assessed mechanical pain thresholds in α-GalA (+/0) (WT) and α-GalA (-/0) (KO) mice treated daily for 30 days with LAC (100 mg/kg) or saline, i.p. (Figure 1 A and B). Measurements were carried out 24 hours and 30 days following ALC withdrawal. In some experiments, the mGlu2/3 receptor antagonist, LY341495 (1 mg/kg) was acutely injected i.p. 30 min before pain measurements. Efficacy of long-term Acetyl-L-carnitine treatment on mechanical allodynia in α-GalA (+/0) (WT) and α-GalA (-/0) (KO) mice. KO mice show an increase mechanical allodynia compare WT mice. Acetyl-L-carnitine (Acetyl-L-carnitine, 100 mg/kg, ip, 30 days) treatment produced a significant increase in PWL (A) and applied force (B) in treated KO mice compared to the saline group (Sal). LY341495 (LY, 1 mg/kg, ip, 30 min prior onset experiment) counteract this effect regarding both PAW (A) and applied force (B). Pharmacological treatments did not induce any effects on WT mice. Data are representative of at least three independent experiments. Values are means ± SEM. Mixed-Way ANOVA followed by LSD Fisher’s post hoc test. *** p < .0001 vs. WT; # p < .05 vs. saline KO; ## p < .01 vs. saline KO; ### p < .001 vs. saline KO.
A mixed ANOVA on latency time of paw withdrawal latency (PWL), with genotype and treatment as grouping factors and sessions (1 and 30 days after the end of treatment, Figure 1(A), left and right graphs, respectively) as dependent variables, showed significant main effect of genotype [F (1, 129) = 86.736, p < .000,001], treatments [F(3, 129) = 12.164, p < .00,001] and interaction genotype*treatment [F(3, 129) = 14.12, p < .000,001]. When we analyzed the within effects (1-day and 30-day after end of treatment), statistical analysis revealed a significant main of session [F (1, 129) = 8.408, p = .00,043). No significance was observed in the interaction session*genotype [F (1, 129) = 0.536, p = 0.46], session*treatment [F (3, 129) = 1.132, p = .33] and session*genotype*treatment [F (3, 129) = 0.88, p = 0.45].
Similar results were observed with regard to the applied force, as shown in figure 1B (main effects: genotype: F(1, 129) = 131,45, p < 0.00,001; treatments F(3, 129) = 13.14, p < 0.00,001; interaction genotype*treatment: F(3, 129) = 16.85, p < 0.00,001; session [F(1, 129) = 35.53, p < 0.0001]; session*genotype [F(1, 129) = 0.56, p = 0.456], session*treatment [F (3, 129) = 1.16, p = 0.327] and session*genotype*treatment [F (3, 129) = 1.52, p = 0.213].
In agreement with our previous data, 32 Fisher’s LSD post hoc test showed that KO mice had a reduction in PWL and applied force compared to WT mice (PWL: 1-day p = 0.000,003; 30-day p = 0.000,004; force: 1-day p = 0.000,001; 30-day p < 0.000,001). Furthermore, no differences between sessions were observed (WTsal/sal 1-day vs WTsal/sal 30-day p = 0.72; KOsal/sal 1-day vs KOsal/sal30-day p = 0.64).
Thirty-day treatment with ALC induced a significant decrease in plantar sensitivity in KO mice to levels similar to those found in WT mice (WTsal/sal vs KOsal/ALC PWL p = 0.51; force p = 0,29; KOsal/sal vs KOsal/ALC PWL p < 0.000,001; force p = 0.000,041). The analgesic effect of ALC was long-lasting because similar results were obtained 30 days after drug withdrawal (WTsal/sal vs KOsal/ALC PWL p = 0.73; force p = 0.6; KOsal/sal vs KOsal/ALC PWL p = 0.000,001; force p < 0.000,001).
ALC-induced analgesia was antagonized by an acute administration of LY341495 (WTsal/sal vs KOLY/ALC PWL p = 0.00,004, force p < 0.000,001; KOsal/sal vs KOLY/ALC PWL p = 0.89, force p < 0.638), which was inactive on its own (KOsal/sal vs KOsal/LY 1-day PWL, p = 0.46; force: p = 0.071).
Of note, neither ALC nor LY341495 treatment induced any effects in WT mice (PWL: sal/sal vs sal/LY341495 p 0.999, sal/sal vs sal/ALC p = 0.9952, sal/sal vs LY341495/ALC p = 0.9878; applied force: sal/sal vs sal/LY341495 p = 0.9981, sal/sal vs sal/ALC p = 0.9999, sal/sal vs LY341495/ALC p = 0.9999).
Effect of 30-day treatment with ALC on thermal hyperalgesia in α-GalA (+/0) (WT) and α-GalA (-/0) (KO) mice
The day after the analysis of mechanical thresholds with von Frey’s filaments, the same mice were subjected to the hot-plate test for the evaluation of thermal hyperalgesia (Figure 2 A and B) by detection of the time of response (latency time) to a thermal cue (52°C ± 1) at day 1 and day 30 after the end of ALC or saline treatment. The effect of a single injection with LY341495 was also evaluated in the hot plate test. A mixed ANOVA on latency time to discomfort reaction, with genotype and treatment as grouping factors and sessions (1 and 30 day after the end of treatment) as dependent variables, showed significant main effect of genotype [F(1, 129) = 20.26, p = 0.00,001], treatments [F(3, 129) = 3.368, p = 0.014] and interaction genotype*treatment [F(3, 129) = 7.282, p = 0.00,014]. When we analyzed the within session effects (1 and 30 day after the end of treatment), there was a significant difference [F (1, 129) = 17.415, p = 0.000,045). No significance was observed in the interaction session*genotype [F(1, 129) = 0.018, p = 0.89], session*treatment [F (3, 129) = 0.587, p = 0.624] and session*genotype*treatment [F (3, 129) = 0.173, p = 0.163]. Efficacy of long-term Acetyl-L-carnitine treatment on thermal hyperalgesia in α-GalA (+/0) (WT) and α-GalA (-/0) (KO) mice. KO mice show an increase in thermal hyperalgesia compared to WT mice. Acetyl-L-carnitine (Acetyl-L-carnitine, 100 mg/kg, ip, 30 days) treatment produced significant increase latency to response to a thermal stimulus in treated KO mice compared to the saline group (Sal). LY341495 (LY, 1 mg/kg, ip, 30 min prior onset experiment) counteract this effect. Pharmacological treatments did not induce any effects on WT mice. Data are representative of at least three independent experiments. Values are means ± SEM. Mixed-Way ANOVA followed by LSD Fisher’s post hoc test. ** p < .001 vs. WT### p < .0001 vs. saline KO.
Fisher’s LSD post hoc test showed that KO mice had a reduction in the latency time compared to WT mice (p = 0.0011). Treatment with ALC induced a significant increase in thermal threshold in KO mice to values similar to those found in WT mice at both 1 day (WTsal/sal vs KOsal/ALC p = 0.46; KO sal/sal vs KO sal/ALC p = 0.000,034) and 30 days (KOsal/sal vs KOsal/ALC p < 0.000,001) after drug withdrawal. Again, the effect of ALC was prevented by a single injection of an acute administration of LY341495. (1-day: p = 0.004; 30-day p = 0.002). LY341495 was inactive on its own (KOsal/sal vs KOsal/LY p < 0.75)
No treatment effects were observed in the WT group (1-day: sal/sal vs sal/LY341495 p = 0.352; sal/sal vs sal/ALC p = 0.282 sal/sal vs LY341495/ALC p = 0.922; 30-day: sal/sal vs sal/LY341495 p = 0.714; sal/sal vs sal/ALC p = 0.212 sal/sal vs LY341495/ALC p = 0.127).
Expression of mGlu2 receptors in cultured DRG neurons
To study whether ALC was able to enhance mGlu2 receptor expression in FD mice, we first assessed mGlu2 receptor protein levels in cultured DRG neurons isolated from α-GalA (+/0) (WT) and α-GalA (-/0) (KO) mice. Using cultured DRG neurons we were able to demonstrated an overexpression of ion channels in FD mice.
35
Hence, this experimental paradigm was considered appropriate for the study of mGlu2 receptors. Mice were sacrificed one day after a 30-day ALC or Saline treatment and DRG neurons were isolated and plated in culture. Total proteins extracts were prepared 48 h after plating, and mGlu2 receptor protein levels were assessed by Western blot using a specific anti-mGlu2 receptor antibody. Immunoblots showed a clear band at approximately 95 kDa, corresponding to the deduced molecular size of receptor monomers (Figure. 3(A), left panel). A significant increase in mGlu2 receptor protein levels was detectable in DRG neurons prepared from ALC-treated KO mice compared to KO mice treated with saline (Figure. 3(A), right panel, p = .0021). A trend to a no-significant increase in mGlu2 receptor expression was also observed in DRG neurons isolated from WT mice treated with ALC as compared to the corresponding group of mice treated with saline ((a), p = .0549). Interestingly, the KO mice treated with saline solution display a decrease in the total expression of the protein, suggesting a possible involvement of the protein in the pathophysiology of the disease. In line with these evidences, we revealed an increase in mGlu2 receptors immunoreactivity in cultured DRG neurons. As shown in Figure 4 panel A, ALC treatment qualitatively increase the fluorescence of mGlu2 receptor immunoreactivity, both in WT e KO DRG and positive for the neuronal marker PGP 9.5. These up-regulation was not long lasting since no increase in protein level nor increase in mGlu2 receptor immunoreactivity was observes in DRG neurons isolated from both genotypes treated with ALC or saline, sacrificed 30 days after the end of the treatment (Figure 3(B), KO SAL vs KO ALC, p = .7534). On the contrary, 2 receptor protein levels and immunofluorescence were significantly lower in DRG isolated from KO mice with respect to WT (Figure 3(B), p = .0069). Western blot analysis of mGluR2 expression in DRG neurons after 30-day ALC treatment at 1 day (A) and 30 days post-treatment (B). A) Western blot analysis of mGluR2 (95.5 kDa) on DRG neurons isolated from 2-month-old mice shows that the protein level is higher in cells prepared from ALC-treated animals in both genotypes compared to saline-injected controls. Statistical analyses of mGluR2 protein expression normalized to β-actin (42 kDa) reveals that such difference is significant comparing the KO-treated and untreated mice. Sal = saline, ALC = acetyl-L-carnitine. B) Western blot analysis of mGluR2 (95.5 kDa) on DRG neurons isolated from 3-month-old mice shows that the protein level is consistently reduced in KO subjects compared to the WT counterpart, regardless of the treatment. Statistical analyses of mGluR2 protein expression normalized to β-actin (42 kDa) reveals that the decline observed in KO cells with respect to WT ones is statistically significant. P < 0.05 (*), p < 0.01 (**). Immunofluorescence analysis of mGluR2 expression in DRGs of 1 day (A) and 30 days (B) after 30-day Acetyl-L-carnitine treatment. The images show a higher immunostaining in DRG cells prepared from Acetyl-L-carnitine-treated animals in both genotypes compared to saline-injected controls, one day after the end of the treatment (A). The effect is not maintained after 30 days of drug withdrawal in cultures DRG (B). Here, WT neurons show a higher mGlu2 receptor signal compared to KO DRG, independently from the treatment. Fluorescent images were captured on a Nikon D-Eclipse C1 inverted laser scanning confocal microscope as single confocal sections at 40X magnification. The EZ-C1 3.90 Free Viewer and Image J (NIH, http://rsb.info.nih.gov/ij/) software were used for image analysis. Scale bar represents 50 µm.).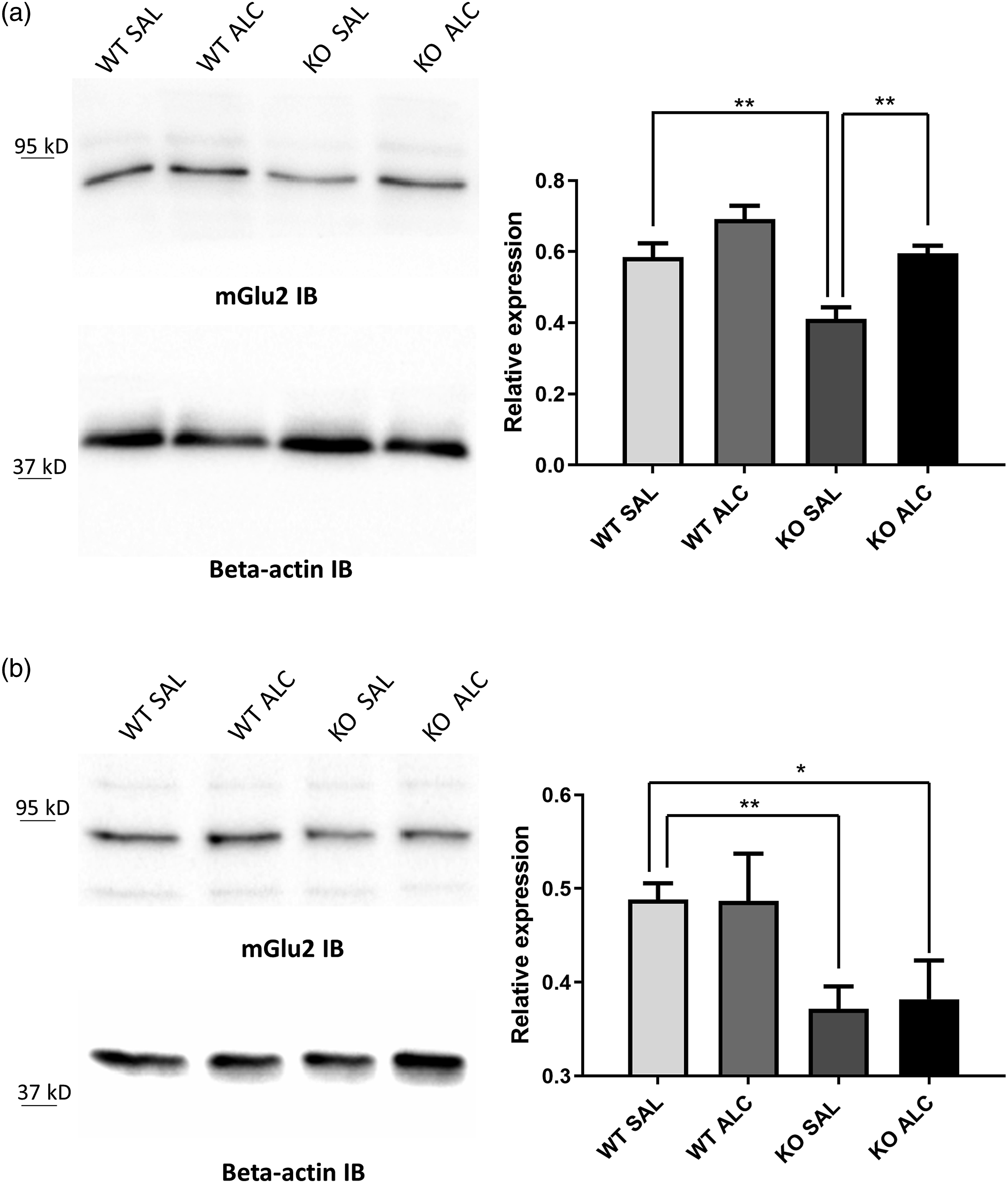

Expression of mGlu2 receptors in the lumbar spinal cord
We performed a qualitatively immunohistochemical analysis of mGlu2 receptors in lumbar (L4-L5) spinal cord sections from WT and KO mice 1 day after treatment with ALC or saline. mGlu2 receptor immunoreactivity was nearly undetectable in laminae I-IV of the dorsal horns of WT and KO mice treated with saline (Figure 5A, B, C), and increased substantially after ALC treatment in both genotypes (Figures 5(D) and (E)). Acetyl-L-carnitine treatment up-regulates mGlu2 receptors in the lumbar spinal cord. A schematic representation of the structures analyzed is shown in (A). Immunohistochemical analysis of mGlu2 receptors in a representative section of the lumbar spinal cord from mice treated with saline or Acetyl-L-carnitine, 1 day post-treatment, is shown in (B,D), and (C,E), respectively. Images show a more intense immunoreactive signal (black arrows) in the dorsal horn, especially in layers 1,2, and 3 (indicated by black hatching in the right horn, panels C,E) of the sections prepared from ALC-treated animals both in WT (C) and KO (E) genotypes compared to saline-injected controls (B and D, WT, and KO, respectively).
Discussion
ALC, an endogenous molecule derived from acetylation of carnitine in the mitochondria, has been extensively studied as a valuable therapeutic option to improve the symptoms and slow the progression of painful neuropathies. 36
The antinociceptive and neuroprotective action of ALC has been demonstrated in a variety of experimental models of neuropathic pain, such as streptozotocin- and chemotherapy-induced neuropathy, chronic constriction injury of the sciatic nerve, and diabetic or HIV neuropaties.24,25,28,37–39 The therapeutic efficacy of ALC has been consistently demonstrated in patients affected by diabetic or HIV painful neuropathy.40–44 ALC causes analgesia by enhancing acetylation of p65/RelA, a member of the Nuclear factor-κB (NF-κB) family of transcription factors.28,45,46 This amplifies Grm2 gene transcription, with a resulting expression of mGlu2 receptors in DRGs and dorsal horns of the spinal cord.28,29,45,46 The mGlu2 receptor, which is coupled to Gi/o proteins, is strategically positioned to restrain glutamate release from sensitized primary afferent sensory fibers. 28 Remarkably, ALC-induced analgesia and up-regulation of mGlu2 receptors persist for a long time after drug withdrawal, perhaps as a result of H3 histone acetylation at Grm2 gene promoter. 29 Thus, ALC can be considered as first-in-class of epigenetic drugs used in the treatment of chronic pain.
Pain is one of the earliest clinical symptoms reported by children and young adults affected by FD, and although some improvement may be obtained by enzyme replacement therapy, pain may still be present, and requires the use of analgesics.47–49 Similarly to patients affected by FD, α-Gal KO mice show mechanical and heat hypersensitivity associated with cold hyposensitivity,32,35 although heat hypersensitivity turns into hyposensitivity with age. 50 We wish to point out that pain-related studies performed on FD mouse models are not homogeneous. Accordingly, two studies found that FD mice show increased sensitivity to radiant heat,50,51 while other studies found decreased sensitivity to heat in these mice.51,52 Contrasting data may arise from the mixed genetic background of the offspring of FD mice, making difficult to obtain strain-matched healthy controls. In our model, we separated homozygous α-Gal KO and WT mice after at least 4 generations and performed all experiments after more than 10 generations, thus eliminating any strain variability in the two genotypes. 33 Our α-Gal KO mice showed clear-cut abnormalities in pain sensation compared to WT mice. These abnormalities are in line with sensory findings obtained in FD patients, in which thermal hyposensitivity has been described in several studies,53–56 and evoked pain with mechanically hypersensitive palmar, and plantar skin is frequently reported. 57 Several clinical studies showed hypersensitivity to mechanical stimuli including allodynia in patients affected by FD,55,58–60 and even the reduced density of cutaneous small nerve fibers found in α−Gla KO mice closely resembles the loss of skin fibers observed in patients affected by FD58,61,62 which might be due to the damaging effect of intracellular Gb3. 63
Our FD mice therefore provide a reliable model for the study of novel therapeutic strategies to relieve pain associated with FD. Here, we have found that a chronic treatment with ALC was able to counteract the higher mechanical sensitivity and restore physiological responses in KO mice. In agreement with previous studies, ALC-induced analgesia appears to be mediated by overexpression and endogenous activation of mGlu2 receptors, since a single injection of the potent mGlu2/3 receptor antagonist, LY341495 (reviewed by 64 ) counteract the effects of ALC in α-Gal KO mice. Noteworthy, mGlu2 receptors are localized along the axons but they are excluded from presynaptic terminals.65,66 Thus, these receptors are not accessible to the glutamate released from nerve endings, unless the onset of receptor-up-regulation, as occurs in response to ALC. This could explain the lack of response of LY341495 alone and its antagonist effect on the ALC-induced analgesia.
Remarkably, ALC-induced analgesia in FD mice persisted at least 1 month after drug withdrawal, similarly to what found in mouse models of chronic inflammatory (CCI) or neuropathic pain 29 and in agreement with the epigenetic mechanism of the drug. Anyway it should be noticed that, although the long-lasting effect of ALC is consistent, ALC up-regulation mGlu2 receptors in the DRGs of α-Gal KO mice appear to be not long lasting since not present after 30 days of withdrawal (in spite of the persistent analgesia and the effect of LY341495 treatment). These results differ from data reported in the CCI model of neuropathic pain, in which an up-regulation of mGlu2 receptors was still observed in the dorsal horns of the spinal cord 37 days after ALC withdrawal. 29 We speculate that in FD mice mGlu2 receptors are transferred from DRGs to nerve endings in the spinal cord 30 days after ALC withdrawal, or, alternatively, that mGlu2 receptors are more functional or becomes more accessible to endogenous glutamate within this timeframe. These hypotheses warrant further investigation.
We believe that these findings are valuable from a translational standpoint, and support the possible use of ALC in order to activate or reinforce the endogenous activation of mGlu2 receptors in the treatment of pain associated with FD. The following aspects should be highlighted:
(1) mGlu2 receptors have been detected in human DRGs, where its activation prevents nociceptive sensitization of sensory neurons 67 ; (2) No tolerance develops to the action of ALC 28 ; (3) ALC is an endogenous compound and has an excellent profile of safety and tolerability (see all references of clinical studies with ALC); (4) ALC has no interaction with cytochrome-P450 or other drug metabolizing enzymes, as opposed to other analgesics used in the treatment of neuropathic pain, such as amytryptiline, duloxetine, or venlafaxine; (5) ALC has been shown to display fast antidepressant effect in experimental animals and humans,68–70 and, therefore, ALC may have the added value to improve depressive symptoms in patients affected by FD; and, (6) the long-lasting effect of ALC might prevent fluctuations in the control of pain. It will be important to examine whether, and to what extent, ALC treatment affects the primary pathological mechanism associated with FD, which is the accumulation of glycosphingolipids as a result of α-Gal deficiency.
Footnotes
Author contributions
F.F and C.D and R.R managed the maintaining of the mice colony, designed and performed all the molecular, behavioral and immunostaining experiments and their analyses, revised critically the manuscript; R.R., R.L revised critically the manuscript and contributed reagents; M.C. and F.N. conceived, designed and supervised the project, wrote the paper. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Fondazione Del Monte di Bologna ID ROL: FdM/4623 and Alfasigma S.p.A Grant number Rep. 62/2020 to M.C. FF was supported by University of Bologna (Assegno di Ricerca 537/2019) and CD is a PhD student of the Department of Pharmacy and Biotechnology.