
Abstract
Extracellular signal-regulated kinases are widely expressed protein kinases in neurons, which serve as important intracellular signaling molecules for central plasticity such as long-term potentiation. Recent studies demonstrate that there are two major forms of long-term potentiation in cortical areas related to pain: postsynaptic long-term potentiation and presynaptic long-term potentiation. In particular, presynaptic long-term potentiation in the anterior cingulate cortex has been shown to contribute to chronic pain-related anxiety. In this review, we briefly summarized the components and roles of extracellular signal-regulated kinases in neuronal signaling, especially in the presynaptic long-term potentiation of anterior cingulate cortex, and discuss the possible molecular mechanisms and functional implications in pain-related emotional disorders.
Keywords
Introduction
The anterior cingulate cortex (ACC) is believed to play a vital role in pain perception, chronic pain, and emotional disorders. Long-term potentiation (LTP) serves as the basic molecular mechanism of synaptic plasticity of chronic pain and pain-related emotions in the ACC.1–4 Previous investigations have reported two forms of LTP in ACC synapses: N-Methyl-D-aspartate receptor (NMDAR)-dependent postsynaptic LTP (post-LTP) and NMDAR-independent presynaptic LTP (pre-LTP).5–7 Notably, pre-LTP is thought to be related to behavioral anxiety, and post-LTP to chronic pain.2,6 Unlike post-LTP, the intracellular mechanism for pre-LTP has been investigated less. In this brief review, we will review previous and recent evidence of the involvement of extracellular signal-regulated kinase (ERK) in synaptic plasticity and discuss possible molecular signaling pathways and functional implications of ERKs in ACC and the insular cortex (IC).
ERK family introduction
ERK is used as a synonym of the mitogen-activated protein kinase (MAPK). ERK1-8 and other signaling molecules, such as c-Jun NH2-terminal kinases 1-3 (JNK1-3), and p38α/β/δ/γ compose of the MAPK family.8–10 MAPKs phosphorylate specific serine and threonine of other protein kinases, phospholipases, transcription factors, and cytoskeletal proteins as substrates. They also play important roles in cell signaling transduction during cell differentiation, cell proliferation, and cell death.
Among all the MAPK family, ERK1 (also named as p44 MAPK) and ERK2 (p42 MAPK) were discovered first.11,12 They can be activated by different biochemical stimuli, such as growth factors, cytokines, virus infection, ligands for G protein–coupled receptors, transforming agents, and carcinogens. Similar with other MAPKs, ERK1/2 are located in a three-kinase phosphorylated cascade that includes the MAPKs, MAPK kinases (MEKs/MAPKKs), and MEK kinases (MEKKs/MKKKs/MAP3Ks). ERK1/2 is activated by a pair of MEKs, MEK1/2. MEK1/2 serves as substrate of Raf isoforms: c-Raf1, B-Raf, or A-Raf, which can be activated by the proto-oncogene Ras. 9 The role of ERK1/2 varies in different cells, whereas both are involved in synaptic activities in central nervous system.
Roles of ERKs in plasticity-related neuronal signaling
In the central nervous system, the ERK signaling pathway is activated by synaptic activity such as membrane depolarization and calcium influx. 13 Activity-dependent activation of NMDARs is known to activate ERKs in hippocampal neurons.14–18 Since NMDARs mediate the induction of LTP, English and Sweatt provided the first evidence that ERK signaling is involved in plasticity of mammalian hippocampus, using the MEK inhibitor PD 98059 to block NMDAR-dependent LTP induction without affecting the NMDAR-mediated transmission. 19 NMDARs also facilitate calcium influx which activates Ref and its downstream ERK cascade. 13 Nitric oxide (NO) may act as the bridge between NMDAR and Ras/ERK cascade. 20
In addition, there are reports of the link from L-type voltage-gated calcium channels (L-VGCCs) to ERKs. Calcium influx through L-VGCC is believed to activate the Res-Ref-ERK cascade, which increase the levels of GTP-bound form of the small G protein Ras. This may be due to the increased activity of guanylyl nucleotide exchange factors, decreased activity of GTPase-activating proteins, or a combination of these two processes. 21 In chick cone photoreceptors, Ras/ERK takes part in the circadian output pathway regulating L-VGCCs, and NO is also involved in this regulating process.22,23
ERKs also have a strong relationship with metabotropic glutamate receptors (mGluRs), especially group I mGluRs (mGluR1/5). Structural studies have proved that ERK1 can bind to mGluR5 in vitro and ERK2 interacts with mGluR1a in rat cerebellum; thus, mGluRs can be phosphorylated and maintain adequate surface expression.24,25 Molecules that activate ERKs, brain-derived neurotrophic factor, for example, elevate mGluR5 phosphorylation in cultured cortical and striatal neurons. This elevation can be blocked by MEK inhibitor, U0126. 26 The activation of group I mGluRs is proved to induce long-term depression (LTD) in the hippocampus and other cortical regions.27–30 ERK1/2 and p38 MAPK have been found to be required for mGluR-LTD.31,32
Moreover, kainate receptors (KARs) also interact with ERKs. In rat striatal slices, superfused KA activated ERK, which can be blocked by applying PD98059. 33 These results were confirmed in cultured striatal neurons. Antagonist of the KAR blocked the activation of ERK. 34 A recent study reported that protein expression of a putative KAR auxiliary subunit Neto2 can be upregulated via MEK/ERK signaling. 35
Postsynaptic modification of AMPAR-mediated responses
Roles of ERK in α-amino-3-hydroxy-5-methyl-4 isoxazolepropionicacid receptor (AMPAR) transmission has been studied for decades. Active Ras leads to increased AMPAR responses, which can be blocked by PD98059. 36 Further studies found that the activation of ERK is required for CaMKII to modulate the insertion of AMPAR into synapses during memory formation. 37 Among all the AMPARs, calcium-permeable-AMPARs (CP-AMPARs) are responsible for activating the Ras/ERK signaling cascade. 38 p38 MAPK is required for the CP-AMPAR mediated LTP in CA1. 39
ERKs and learning-related hippocampal LTP
One key function of ERK and its cascade is in the synaptic plasticity. The ERK signaling pathway might regulate synaptic targets to control important functions such as synaptic plasticity, learning and memory in the adult brain. Theta burst stimulation (TBS) induces LTP in the hippocampus through calpain-1-mediated suprachiasmatic nucleus circadian oscillatory protein degradation, ERK activation, and actin polymerization. 40 By using fluorescence resonance energy transfer (FRET)-based sensors, the activation of ERK during structural LTP can be detected. 41 In the mouse hippocampus, an increase in nuclear Poly(ADP-ribose) polymerase-1 and Poly(ADP) ribose molecules during the Foskolin-induced LTP is regulated by the activation of cAMP-dependent protein kinase (PKA) and ERK.42–44
Anterior cingulate cortex LTPs
Previous studies have reported that LTP exist in cortical areas that are important for pain perception.1,3 Table 1 summarizes different types of ERKs and their roles in LTP in the cortical areas. Among all the cortical areas, pharmacological and genetic studies reveal that LTP in the ACC exists in at least four different forms: NMDAR-dependent, L-VGCC-dependent, late-phase LTP (L-LTP), and pre-LTP.2–4 The first is NMDAR-dependent LTP. Application of the NMDAR antagonist AP-5 blocked the induction of LTP. Both NMDARs containing GluN2A (called NR2A) or GluN2B (NR2B) subunits contribute to this form of LTP. 5 Second is calcium channel-dependent LTP. L-VGCCs are required for the induction of LTP by TBS in the field recording condition in the ACC. Unlike LTP recorded using field recording, LTP recorded using whole-cell patch clamp does not respond to the inhibition of L-VGCCs. 45 Third is late-phase LTP. Recent studies using a 64-channel multi-electrode array (MED64) show that ACC LTP induced by multiple TBS can last more than 5 h. 7 This form of potentiation is sensitive to inhibition of protein synthesis, indicating that protein synthesis-dependent L-LTP exists in the ACC. 46 It is likely that L-LTP may contribute to long-term changes in the cortical circuits that are triggered by peripheral injury. It is believed that all these three forms of LTP are expressed postsynaptically, since they are all sensitive to genetic deletion of AMPAR subtypes or inhibition of AMPAR subtypes.
Cortical ERKs in different forms of LTP.
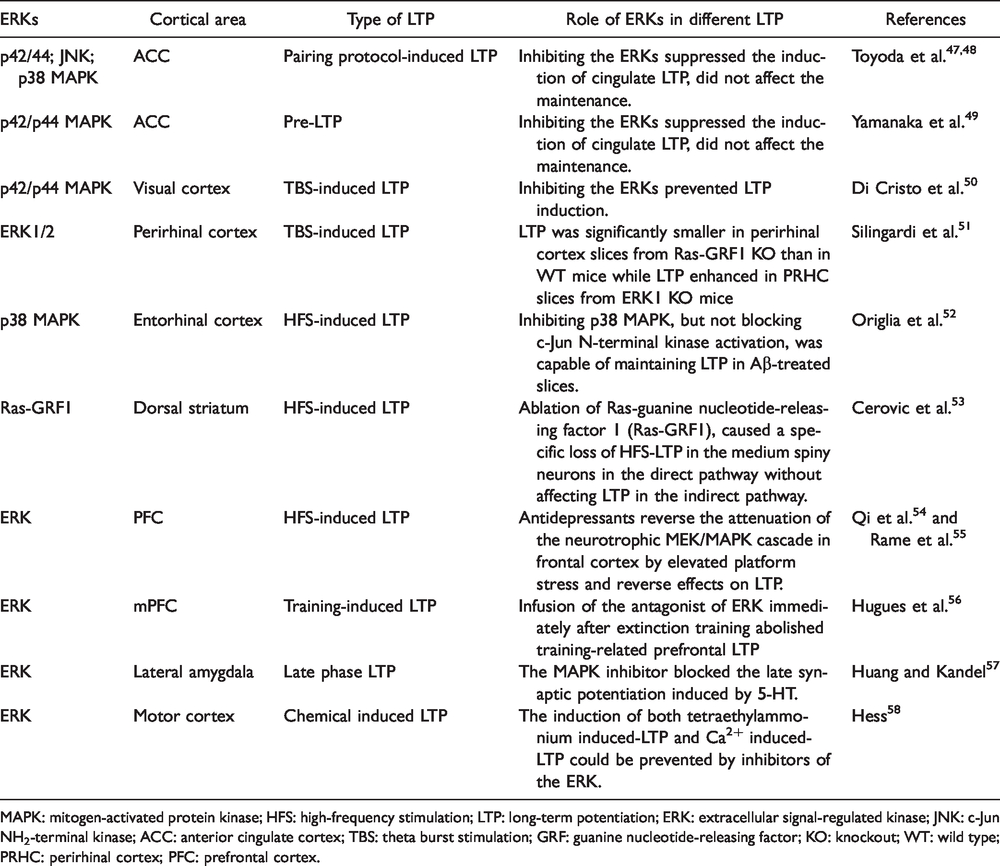
MAPK: mitogen-activated protein kinase; HFS: high-frequency stimulation; LTP: long-term potentiation; ERK: extracellular signal-regulated kinase; JNK: c-Jun NH2-terminal kinase; ACC: anterior cingulate cortex; TBS: theta burst stimulation; GRF: guanine nucleotide-releasing factor; KO: knockout; WT: wild type; PRHC: perirhinal cortex; PFC: prefrontal cortex.
Our recent study reported that a new pairing protocol can induce LTP in the ACC that is purely presynaptically induced. The induction of pre-LTP is NMDAR-independent, a key feature differentiating pre- from post-LTP in the ACC. 6 Glutamate KA GluK1 receptor is important for the induction of pre-LTP. This was nicely demonstrated by the use of GluK1 knockout mice as well as a selective GluK1 inhibitor. The requirement of GluK1 is subtype selective, since GluK2 is not required for pre-LTP. In addition, we found that L-type voltage-gated calcium channels can also contribute to the induction of pre-LTP, since nimodipine reduced the amplitude of pre-LTP.
ERKs in post-LTP in ACC and IC
Electrophysiological studies show that LTP induced in ACC was completely blocked by PD98059 and U0126 applied postsynaptically. 47 The inhibitory effect is selective for the induction of post-LTP, and the MEK inhibitors did not affect the maintenance of cingulate LTP (Figure 1). Inhibitors of JNK, and p38 MAPK, SP600125, and SB203850, also suppressed the induction of cingulate LTP generated by the pairing protocol. These results suggest that the activation of MAPK including ERK, JNK, and p38 MAPK, is critical for the induction of LTP in the ACC. Furthermore, activated ERK/MAPK likely has multiple targets including cAMP response element-binding protein (CREB), which is required for long-term synaptic changes in neurons. 59 A previous study also reported that GluA1−/− mice showed a significant decrease in phosphorylation of ERK1/2 in neurons of the ACC in animal models of inflammatory pain, 48 which indicated that GluA1 may act upstream of ERK.
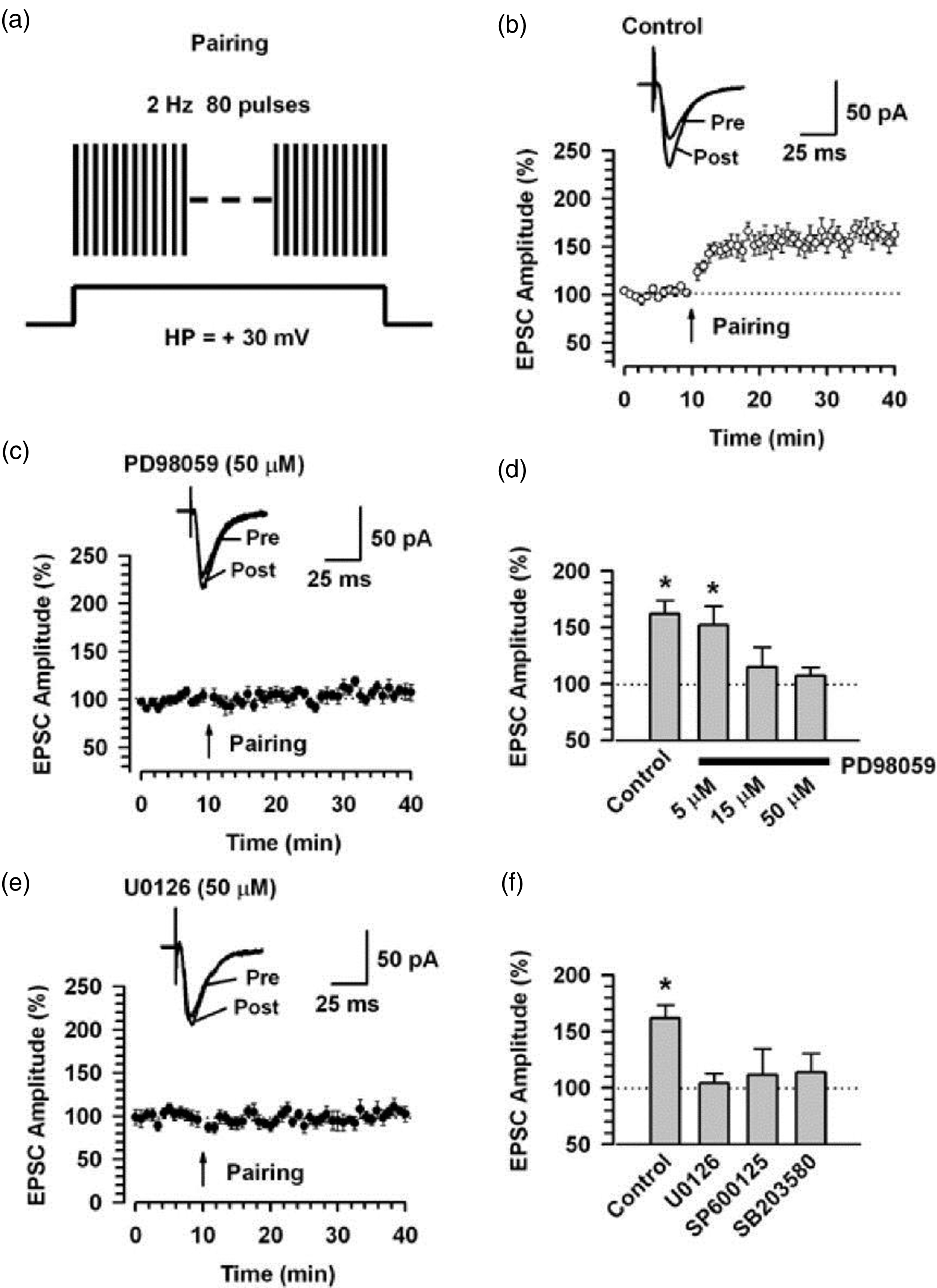
Requirement of ERKs for cingulate post-LTP. (a) A scheme illustrating the LTP induction protocol consisting of pairing 80 presynaptic pulses at 2 Hz with postsynaptic depolarization (holding at + 30 mV). (b) LTP is induced by the pairing protocol in cingulate pyramidal neurons (n = 13). (c) The MEK inhibitor, PD98059 (50 µM, n = 10) in the intracellular solution completely blocks LTP induction. Traces show averages of six EPSCs at baseline response (pre) and 30 min (post) after the pairing protocol (arrow). The dashed line indicates the mean basal synaptic response. (d) Summary of the effects of PD98059 at different concentrations on LTP induced by the pairing protocol (PD98059; 5 µM, n = 8; 15 µM, n = 6, 50 µM, n = 10). *P < 0.05 compared to baseline response. (e) The MEK inhibitor U0126 (50 µM) in the intracellular solution completely blocks LTP induction. Traces show averages of six EPSCs at baseline response (pre) and 30 min (post) after the pairing protocol (arrow). The dashed line indicates the mean basal synaptic response. (f) Summary of the effects of MAPK inhibitors on LTP induced by the pairing protocol. *P < 0.05 compared to baseline response (adapted from Toyoda et al. 47 ).
There are reports of post-LTP in the IC;60,61 however, there have been no studies of ERKs requirements for post-LTP yet. A previous study using in vivo field recording reported that ERK activity is required for LTP of excitatory responses evoked by focal stimulation in the amygdale. 62 However, it is unclear if that form of LTP is post-LTP.
ERKs are required for the induction, but not expression, of pre-LTP
Pre-LTP has been investigated in ACC and IC so far, and the induction of pre-LTP in ACC was found to block by the MAPK and MEK inhibitors 49 (Figure 2). By contrast, the MEK inhibitors did not affect the maintenance of ACC pre-LTP. These results suggest that the activation of MAPK including ERK is critical for the induction of pre-LTP in the ACC and indicate that ERK inhibitors may affect mood-related behaviors by inhibiting pre-LTP in the ACC.

Requirement of ERKs for cingulate pre-LTP. (a) A scheme illustrating the pre-LTP induction protocol consisting of low-frequency paired-pulse stimulation (2 Hz for 2 min) at a holding potential of −60 mV. (b) Top: sample traces of eEPSCs with paired pulse stimulation at 50 ms interstimulus interval during baseline (1) and 60 min after the pre-LTP (2) at a holding membrane potential of −60 mV. Middle: a time course plot of a representative single example. Bottom: time course plot of PPR for this neuron. The arrow donates the time of pre-LTP induction. (c) Pooled data to illustrate the time course of pre-LTP and changes in PPR. Top: pre-LTP (red, n=7 neurons/5 mice) and control (gray, n=6/4). Bottom: PPR for these neurons. *P<0.05 for PPR change estimated for the 10-min intervals baseline and between 50 min and 60 min after the induction stimulation. (d) Top: a time course plot of a representative single example with application of ERK inhibitor, PD98059 (100 mM). Bottom: time course plot of PPR for this neuron. (e) PD98059 (blue, n=10/6) in the bath solution blocks pre-LTP induction. (f) Summary of the effects of an ERK inhibitor, PD98059 on pre-LTP. There was statistical difference when comparing control with pre-LTP, PD98059 groups. The mean amplitudes of eEPSCs were determined at 50–60 min after the pre-LTP induction stimulation (adapted from Yamanaka et al. 49 ).
Other key molecules that are necessary for the induction of pre-LTP have been discovered in the ACC (Figure 3). The activation of GluK1 containing KARs and L-VGCC are necessary. One key protein kinase involved in pre-LTP is PKA. 6 However, it has been reported that presynaptic MAPK is activated by cAMP independently of PKA. 63 In neurons of rat prefrontal cortex (PFC), cAMP activates ERK signaling pathways to induce presynaptic potentiation. Recent studies reported that the loss of the fragile X mental retardation protein (FMRP), which is the product of the Fmr1 gene, causes the failure to induce pre-LTP in the ACC. 64 The translocation of PKA subunits in Fmr1 knockout mice might cause changes in PKA activity, thus resulting in the loss of pre-LTP in the ACC. SCRAPPER, a major presynaptic E3 ubiquitin ligase, also participates in the induction of pre-LTP in the ACC by manipulating the uptake of glutamate. 65 This effect is also considered as one result of PKA activation. Recent studies have also discovered pre-LTP in the IC, 66 but no study is available about the role of ERKs in pre-LTP.

Model for signaling pathways of ERKs in pre- versus post-LTP in cortex. In presynaptic terminals, ERK acts as the downstream of AC1 in a Ca2+-CaM dependent manner. ERKs phosphorylate synapsin 1 thus facilitate the glutamate release. Inhibitors of MEK block the induction of pre-LTP without affecting the maintenance. Molecules such as PKA, FMRP, SCRAPPER and HCN channels have been reported to play key roles in pre-LTP. In post synaptic terminals, inhibitors of ERK block the induction of NMDAR-dependent and independent LTP in ACC. PKA activates ERKs via the activation of AC1. Activated ERK targets multiple molecules thus cause the long term synaptic changes. GluA1 may also act upstream of ERK in the ACC.
AC1 may act upstream from ERKs in cortical LTPs
Adenylyl cyclase 1 (AC1), an adenylyl cyclase subtype that is activated by Ca2+-CaM, has been reported to play essential roles in central synaptic plasticity.59,67,68 Our previous studies have reported that AC1 is a key upstream enzyme for the activation of ERKs in spinal dorsal horn neurons by peripheral injury. 69 In genetic knock-out mice of AC1, the activation of ERK in dorsal horn neurons were significantly reduced or blocked, either after peripheral tissue inflammation or by glutamate or substance P in spinal cord slices. In the hippocampus, the overexpression of AC1 elevated the expression of phosphorylated ERK1/2, 70 supporting the idea that AC1 may act upstream from ERKs. In the ACC, AC1 is known to contribute to both pre- and post-LTP. In an animal model of chronic visceral pain, it has been found that the expression of AC1 was upregulated in the ACC. 71 It is likely that AC1 may be located both presynaptically and postsynaptically.6,68 A recent study using cultured cortical neurons shows that the transcriptome of AC1 is found in excitatory presynaptic terminals. 72
Possible molecular mechanisms for ERKs in pre-LTP
The activation of ERK was reported to facilitate glutamate release from synaptosomes in rat brain by phosphorylating the synaptic vesicle membrane protein synapsin I, thereby regulating its interaction with the actin cytoskeleton and leading to the recruitment of releasable synaptic vesicles from a distal pool.73,74 PD98059 and U0126 inhibited pre-LTP in the ACC, indicating that the activation of MAPK/ERK may contribute to enhanced release of glutamate in ACC pre-LTP. 49 Maintenance of memory-related long-term facilitation of presynapses requires the upregulation and prion-like activation of CREB, a synaptic translational regulator through MAPK/ERK signaling. However, the maintenance of cingulate pre-LTP was not affected by both PD98059 and U0126. This suggests that the MAPK/ERK signaling cascade is not persistently activated during pre-LTP in the ACC.
It has been reported that glutamate releasable vesicles are increased by ERK.73,75 Presynaptic vesicle mobilization is a complex phenomenon that is regulated by a number of protein kinases. Previous studies have demonstrated that ERK can increase releasable vesicles and induce glutamate exocytosis by phosphorylation of synapsin I, which is a major substrate for ERK and a presynaptic protein regulating the vesicle cycle and neurotransmitter release. Although synapsin I anchors synaptic vesicles to cytoskeletal elements under the inactive condition, once phosphorylated by ERK, it dissociates from synaptic vesicles and increases more releasable vesicles at presynaptic active zone for neurotransmitter release. ERK/MEK inhibitors did not affect baseline pair-pulse ratio and spontaneous excitatory postsynaptic current (EPSC) in ACC neurons under resting conditions. This means that these kinases can be activated by a condition such as low-frequency stimulation and then enhance glutamate releases on presynaptic neurons.
Acute pain versus chronic pain
Previous studies have reported that ERKs were upregulated after peripheral tissue or nerve injury.48,69,76–79 For example, acute intraplantar injection of formalin induced rapid activation of ERKs in some of layer II neurons in the bilateral ACC, while non-noxious stimulation was not able to significantly activate ERKs (Figure 4). 69 Furthermore, in animal models of neuropathic pain and chronic inflammation, ERKs in the ACC were significantly upregulated.48,76 Interestingly, after peripheral injury (digit amputation), ERKs can be activated by allodynic non-noxious stimulation. 69 The activation of ERKs has been also reported in other cortical areas such as IC, PFC, and prelimbic cortex in similar chronic pain models.77–79

Enhanced ERK activation in the ACC after tissue and nerve injury. (a) Immunohistochemical staining for phosphorylation of ERK illustrated time course-dependent activation of ERK in layer II neurons of the contralateral ACC after unilateral hindpaw injection of formalin (5%, 50 µl, n = 4–5 rats for each time point). (b) The phosphorylated ERK expression in the layer II ACC neurons and their main apical dendrites (arrows) was increased at two weeks after the amputation of the unilateral hindpaw third digit (n = 5), compared to sham animals (n = 3). (c). Mechanical stimulation by brushing hindpaw of digit amputation induced Perk expression in more number of layer II ACC neurons and the more distinctive apical dendrites at two weeks after the amputation (n = 5), compared to that in rats with amputation alone. There was not phosphorylated ERK activation in the ACC in normal animals after the brushing (n = 3). Left and middle columns: low power of the coronal ACC sections. Scale bar = 50 µm; Right column: enlarged layer II regions corresponding to the small rectangle areas in the middle column, respectively. Scale bar = 25 µm. (adapted from Feng Wei and Zhuo 60 ).
Physiological and pathological significance
Since pre-LTP has been shown to contribute to behavioral anxiety, especially chronic pain-related anxiety, this study provides a possible explanation that ERKs may contribute to mood control by triggering pre-LTP in ACC pyramidal cells. Behavioral studies indicate that pre-LTP is related to anxiety induced by chronic pain. 6 Inhibiting the expression of pre-LTP by hyperpolarization-activated cyclic nucleotide gated (HCN) channel inhibitors in the ACC reduced enhanced anxiety-like behaviors in animals with chronic pain. Post-LTP, which is important for behavioral sensitization, was not affected by HCN channel inhibition. Pre-exposure to a stimulus that causes anxiety can reduce the probability that synapses undergo pre-LTP in brain slices, further suggesting that pre-LTP is involved in anxiety. These results also suggest that pre-LTP may serve as one of the general mechanisms for heighted anxiety, in addition to anxiety specifically caused by chronic pain.
In summary, recent studies have provided new insights into basic mechanisms underlying chronic pain and chronic pain related anxiety. Long-term synaptic changes can take place in both presynaptic and postsynaptic terminals, which are triggered and maintained by a series of activity-dependent signaling molecules. The ACC, along with other less-studied cortical regions, such as the IC, has a crucial role in the affective component of chronic pain. As one kind of important phosphor-regulating effectors, ERK plays essential roles in the signaling pathway in both pre- and post-LTP. The association of pre-LTP with chronic pain-induced anxiety may provide potential opportunities to identify novel drug targets for chronic anxiety.
Footnotes
Acknowledgment
The authors would like to thank Melissa Lepp for English editing.
Author Contributions
QYC, PYX, and MZ drafted the manuscript and finished the final version of the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.