
Abstract
Background
The major dose-limiting toxicity of paclitaxel, one of the most commonly used drugs to treat breast cancer, is peripheral neuropathy (paclitaxel-induced peripheral neuropathy). Paclitaxel-induced peripheral neuropathy, which persists into survivorship, has a negative impact on patient’s mood, functional status, and quality of life. Currently, no interventions are available to treat paclitaxel-induced peripheral neuropathy. A critical barrier to the development of efficacious interventions is the lack of understanding of the mechanisms that underlie paclitaxel-induced peripheral neuropathy. While data from preclinical studies suggest that disrupting cytoskeleton- and axon morphology-related processes are a potential mechanism for paclitaxel-induced peripheral neuropathy, clinical evidence is limited. The purpose of this study in breast cancer survivors was to evaluate whether differential gene expression and co-expression patterns in these pathways are associated with paclitaxel-induced peripheral neuropathy.
Methods
Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology were identified between survivors who received paclitaxel and did (n = 25) or did not (n = 25) develop paclitaxel-induced peripheral neuropathy.
Results
Pathway impact analysis identified four significantly perturbed cytoskeleton- and axon morphology-related signaling pathways. Weighted gene co-expression network analysis identified three co-expression modules. One module was associated with paclitaxel-induced peripheral neuropathy group membership. Functional analysis found that this module was associated with four signaling pathways and two ontology annotations related to cytoskeleton and axon morphology.
Conclusions
This study, which is the first to apply systems biology approaches using circulating whole blood RNA-seq data in a sample of breast cancer survivors with and without chronic paclitaxel-induced peripheral neuropathy, provides molecular evidence that cytoskeleton- and axon morphology-related mechanisms identified in preclinical models of various types of neuropathic pain including chemotherapy-induced peripheral neuropathy are found in breast cancer survivors and suggests pathways and a module of genes for validation and as potential therapeutic targets.
Keywords
Introduction
Chronic chemotherapy-induced peripheral neuropathy (CIPN) is one of the most common adverse effects of neurotoxic chemotherapy (CTX), with prevalence rates that range from 30% to 70% in cancer survivors. 1 While initially described as a reversible condition, a growing body of evidence suggests that CIPN persists long into survivorship. 2 In fact, in 2014, CIPN was added to the National Comprehensive Cancer Network’s Clinical Practice Guideline for Survivorship3,4 because of its substantial negative impact on survivors’ functional status and quality of life.5–10
Paclitaxel is one of the most commonly used neurotoxic drugs to treat breast cancer. 11 While it is well known to stabilize microtubules with resultant impairments in axonal transport, a recent review suggested that the pathophysiologic mechanisms for paclitaxel-induced peripheral neuropathy (PIPN) extend beyond microtubule impairment 12 and may involve mitochondrial damage13,14 and alterations in immune function. 15 We recently described perturbations in mitochondrial dysfunction-related pathways that were associated with PIPN in a sample of breast cancer survivors. 13
The anti-tumor activity of paclitaxel occurs as a result of its ability to alter normal regulatory mechanisms that control microtubule dynamics and microtubule-based transport that are required for a cell’s survival.16,17 Preclinical evidence suggests that the administration of paclitaxel results in microtubule polymerization and stabilization in cancer cells. 18 Within peripheral neurons, paclitaxel binds to β-tubulin in polymerized microtubules, which causes a conformational change and renders these microtubules less dynamic.18,19 Microtubule hyper-stabilization primarily occurs on the most distal portion of the axon and may contribute to the “dying back” phenomenon associated with PIPN.20–24 Recent preclinical evidence suggests that the neurotoxic effects associated with the stabilization of microtubules results in defects in axonal transport,25,26 as well as morphological (e.g., increase in myelin abnormalities,17,27 increase in number of Schwann cell nuclei17,27), and biochemical (e.g., long-term retention of paclitaxel) changes in peripheral nerves.17,27 The administration of paclitaxel results in the degradation of both peripheral and central branches of dorsal root ganglia (DRG) neurons. 28 The mechanisms mediating axonal degeneration in PIPN remain an area of active investigation (reviewed in the study by Fukuda et al. 24 ), providing new opportunities for therapeutic interventions. 29 Given the preclinical evidence that paclitaxel alters cytoskeleton structure and axon morphology and that these alterations are associated with the development and maintenance of peripheral neuropathy, we provide evidence to support the suggestion that perturbations in signaling pathways and patterns of gene co-expression associated with cytoskeleton and axon morphology are also associated with chronic PIPN in breast cancer survivors.
Materials and methods
Survivors and settings
The methods for this analysis, which is part of a larger study of CIPN, are described in detail elsewhere. 10 In brief, survivors were recruited from throughout the San Francisco Bay area and met pre-specified inclusion and exclusion criteria. The National Coalition for Cancer Survivorship’s definition of a cancer survivor (i.e., a person is a cancer survivor from the moment of diagnosis through the balance of life) was used in this study. 30 Of the 1450 survivors who were screened, 754 enrolled, and 623 completed the self-report questionnaires and study visit. Data from a randomly selected sample of breast cancer survivors with (n = 25) or without (n = 25) chronic PIPN were used in this analysis.
Study procedures
Research nurses screened and consented the survivors over the phone; sent and asked them to complete the self-report questionnaires prior to their study visit; and scheduled the in-person assessment. At this assessment, written informed consent was obtained, responses to questionnaires were reviewed for completeness, and objective measurements were obtained. Blood samples were drawn, processed, and stored for subsequent molecular analyses in PAXgene® Blood RNA tubes (Qiagen, Inc.). This study was approved by the Institutional Review Board of the University of California, San Francisco.
Study measures
Demographic and clinical characteristics
Breast cancer survivors provided information on demographic characteristics and completed the Alcohol Use Disorders Identification Test (AUDIT), 31 Karnofsky Performance Status (KPS) scale,32–34 and the Self-Administered Comorbidity Questionnaire.35,36
Pain measures
Survivors with PIPN rated their pain intensity using a 0 to 10 numeric rating scale and completed the pain interference scale from the Brief Pain Inventory 37 and the Pain Quality Assessment Scale. 38
Acquisition and processing of gene expression data
In this study, we used two different but complementary approaches to evaluate whole-transcriptome data for patterns of gene expression associated with PIPN in pathways associated with cytoskeleton and axon morphology (Figure 1). The first approach utilized pathway impact analysis (PIA) in which pre-defined pathways are evaluated for perturbations using the magnitude and significance of gene–gene interactions from differential gene expression (DGE) data. The second approach (i.e., weighted gene correlation network analysis (WGCNA)) constructs networks of genes with shared patterns of co-expression agnostic of phenotypic characteristics, including PIPN. These networks were then evaluated for association with chronic PIPN and for biological processes related to cytoskeleton and axon morphology.

An overview of the two analytic approaches used to evaluate for cytoskeleton- and axon morphology-related gene (G) expression patterns associated with paclitaxel-induced peripheral neuropathy in breast cancer survivors (S) with (C) and without (N) PIPN: [1] PIA and [2] WGCNA. The KEGG pathways and Gene Ontology categories were evaluated for cytoskeleton and axon-morphology biological processes. DGE: differential gene expression; KEGG: Kyoto Encyclopedia of Genes and Genomes; MLR: multiple logistic regression; PIA: pathway impact analysis; PPI: protein–protein interaction; WGCNA: weighted gene co-expression network analysis.
The methods for the quantification of gene expression are described in detail elsewhere. 13 Gene expression of total RNA, isolated from peripheral whole blood, was assayed using RNA-seq. Gene expression was summarized as counts per gene and used as input for the PIA and WGCNA.
PIA of the whole transcriptome
The DGE was quantified using a general linear model as previously described. 13 These DGE analyses were adjusted for demographic (i.e., age, employment status) and clinical (i.e., AUDIT score, body mass index (BMI), KPS score) characteristics that differed between the PIPN groups, as well as for technical variability (e.g., potential batch effects). We estimated 13 that, at a type I error rate of 0.01, we were powered 39 to detect 1.5-fold changes for 83% of genes.
The DGE results were summarized as the log fold change and p value for each gene. PIA was used to evaluate for perturbations in well-defined signaling pathways as previously described. 13 PIA is a topology-based approach to pathway analysis (reviewed in the studies by Khatri et al. 40 and Garcia-Campos et al. 41 ). Unlike other approaches, PIA is not limited by a pre-defined set of genes, does not assume independence of genes, and pathways are considered independent. PIA includes potentially important biological factors (e.g., gene–gene interactions, flow signals in a pathway, pathway topologies), as well as the magnitude (i.e., log fold-change), and p values from the DGE analysis (reviewed in Mitrea et al. 42 ). This PIA included the results of the DGE analysis for all genes (i.e., cutoff free) to determine the probability of pathway perturbations (pPERT) using Pathway Express. 43 This pathway-topology impact factor analysis approach is widely used with over 1200 citations to date. 44
A total of 208 signaling pathways were defined using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. 45 Sequence loci data were annotated with Entrez gene IDs. The gene names were annotated using the Human Genome Organisation (HUGO) Gene Nomenclature Committee resource database. 46 We assessed for significance of the PIA using a strict false discovery rate (FDR) of 1 under the Benjamini–Hochberg (BH) procedure.47,48 Finally, we evaluated these results for pathways related to cytoskeleton and axon morphology.
Gene correlation network analysis
One limitation of PIA is its dependence on pre-defined signaling pathways. By evaluating whole-transcriptome level data (i.e., not filtered by pathway) across multiple samples, patterns of gene co-expression can be identified. Gene co-expression can be identified as sets of genes (i.e., modules) which share more similar expression patterns to each other than they do to other genes in the dataset. These co-expression groups of genes are identified independent of an outcome (e.g., PIPN status) or biological organization (e.g., pathways) to provide an independent interpretation of gene expression data. These co-expression modules tend to be functionally related and co-regulated and if associated with an outcome of interest (i.e., PIPN group) may present new insights into its molecular biology. Because we were interested in identifying novel patterns of gene expression, we generated a co-expression gene network and evaluated this network for associations with chronic PIPN. 49 First, we identified modules that demonstrated co-expression patterns in our dataset. Second, we evaluated these modules for associations with chronic PIPN group membership. Third, we evaluated any module associated with chronic PIPN group membership for patterns of higher orders of biological organization. Finally, we evaluated these results for pathways related to cytoskeleton and axon morphology.
Gene level summaries of RNA abundance were estimated as the log transformed reads per mean thousand (i.e., log2(RPMK + 1)). Modules of genes with highly correlated expression were identified from the top 5000 most variant genes using WGCNA.50,51 We selected an empirical soft power threshold of 11, which represented a strong model fit to a scale free topology (signed R2 = 0.80) to generate the signed adjacency matrix. A clustered gene tree was generated using the “average” method. The gene tree was dynamically cut (with WGCNA parameters: minModuleSize =20, method=“hybrid”, and deepSplit = 2) and merged with a dissimilarity threshold of 0.8 to construct the merged modules. Each module was assigned a color, and this color label was used for identification in all subsequent analyses. Although the performance of co-expression network analysis is dependent of sample size, WGCNA performs well on sample sizes of >20.52,53
Multiple logistic regression analysis, which controlled for significant demographic (i.e., age, employment status) and clinical (i.e., AUDIT score, BMI, KPS score) characteristics, was used to evaluate for an association between the PIPN group and the eigengenes (i.e., the first principal component of variation among the co-expressed genes in that module) of each of the co-expression modules. A backward stepwise approach was used to create a parsimonious model. The binomial regression and the backward stepwise method were performed using the “glm()” and “step()” functions in the “stats” package of R. We assessed for significance of the regression analyses using a p value of <.05.
Functional analysis of the gene correlation network modules
To evaluate for higher orders of biological organization of genes in a module associated with PIPN, we utilized two different approaches (Figure 1). First, we performed a functional enrichment analysis of gene ontology (GO) annotations54,55 and KEGG pathways using ToppFunn and all of the genes in the brown module. 56 We assessed for significance using an FDR of 5 under the BH procedure.47,48 Second, we evaluated connectivity among differentially expressed genes (DEG) in a given module and evaluated for functional enrichment of Reactome pathways 57 using Search Tool for the Retrieval of Interacting Genes (STRING). 58 DEGs were identified at an FDR of 10 under the BH procedure.47,48 We assessed for significance of the DEG protein–protein interaction (PPI) network enrichment using a p value of <.05. We also assessed for functional enrichment of DEGs in the Reactome pathways using an FDR of 10 under the BH procedure.47,48
Results
Differences in demographic, clinical, and pain characteristics
Sample characteristics were reported previously. 13 In brief, breast cancer survivors with chronic PIPN were significantly older (p = .006) and were more likely to be unemployed (p = .022) (Supplementary Table 1). In terms of clinical characteristics, survivors with PIPN had a lower AUDIT score (p = .012), a higher BMI (p = .011), and a lower KPS score (p < .001) (Supplementary Table 2). Of note, no between-group differences were found in the total dose of paclitaxel received or in the percentage of patients who had a dose reduction or delay due to PIPN. The worst pain severity was reported as 6.3 (±2.1) and the duration of PIPN was 3.8 (±3.9) years (Supplementary Table 3).
PIA of the whole transcriptome
Of the 11,487 genes identified in the DGE analysis, 11,174 unique genes were successfully annotated with Entrez IDs and included in the PIA. Fifty-three KEGG signaling pathways were significantly perturbed between the PIPN groups at a strict FDR of 1. 13 Of these 53 pathways, four were associated with cytoskeleton and axon morphology (Figure 1 and Table 1).
Differential perturbation of pathways associated with cytoskeleton and axon morphology in PIPN.

FDR: false discovery rate; PIPN: paclitaxel-induced peripheral neuropathy; totalPert: total perturbation score; totalPertNorm: total normalized perturbation score; pPert: p value of perturbation; HIF-1: hypoxia-inducible factor 1.
Gene co-expression network analysis
Using WGCNA, gene co-expression profiling of the 5000 genes with the most variable expression clustered into three modules (i.e., brown, n = 1207; green, n = 1726; dark red, n = 2067; Figure 2). To evaluate the association between PIPN group membership and each of these co-expression modules, we fit multiple logistic regression models to predict the PIPN group membership using the module eigengenes, AUDIT score, BMI, and KPS score. Of these three modules, PIPN group membership was significantly associated only with the brown module’s eigenenes (Figure 1 and Table 2).
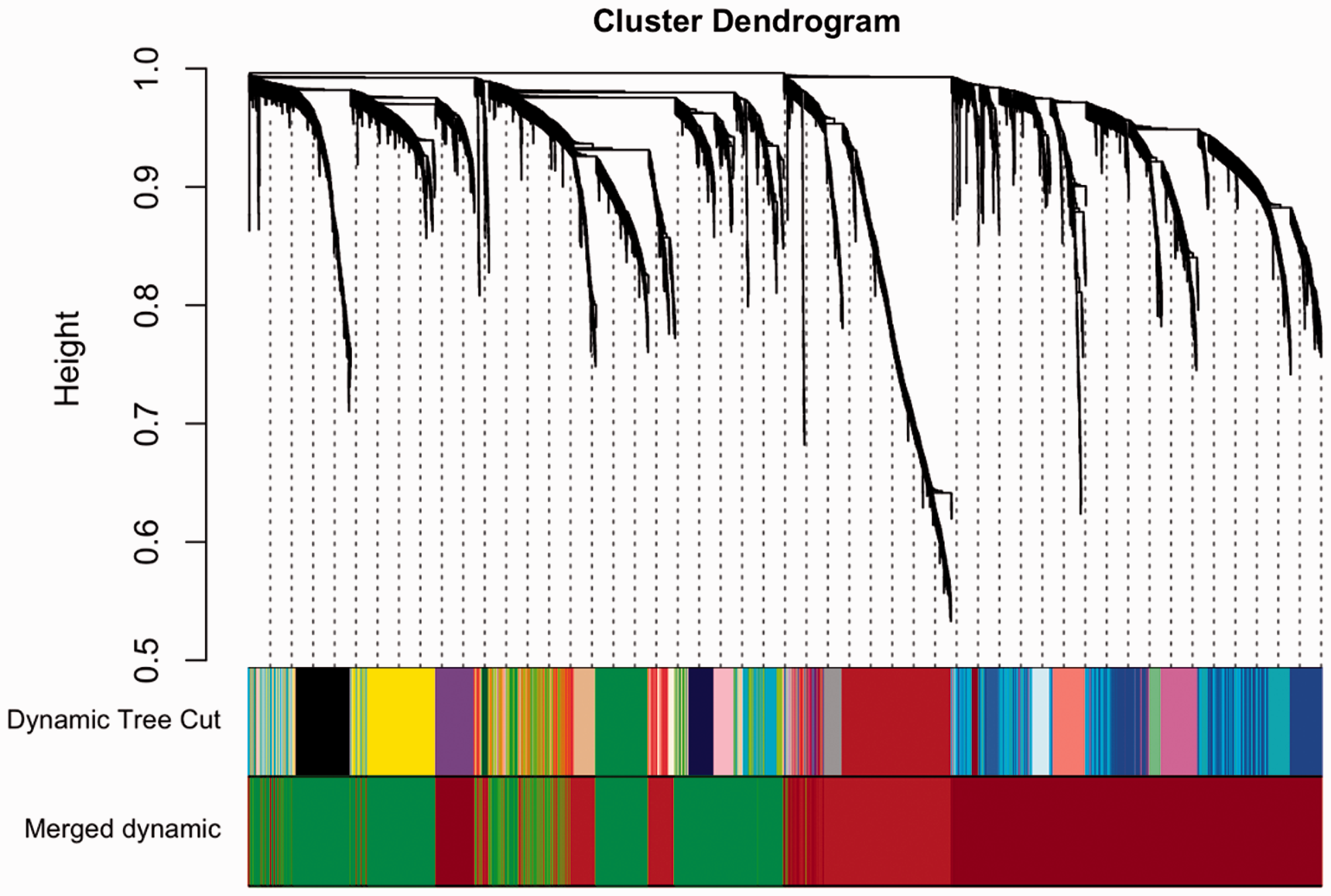
Cluster dendrogram and module colors showing the module assignments for the gene co-expression data. The color rows underneath the cluster dendrogram show the module assignment from the unmerged Dynamic Tree Cut method and the final module set from the Merged dynamic method.
Multiple logistic regression analysis for the brown co-expression module and PIPN.

CI: confidence interval; KPS: Karnofsky Performance Status; r: Pearson’s product moment correlation coefficient; PIPN: paclitaxel-induced peripheral neuropathy.
In our functional analyses of the brown module using ToppFunn, 49 GO annotations (n = 10 biological processes, n = 36 cellular component, n = 3 molecular function) and 27 KEGG signaling pathways were found that were significantly enriched for genes in this module. Two GO categories and three KEGG pathways were related to cytoskeleton and axon morphology (Table 3). To evaluate for enrichment of PPI of DEGs in the brown module, we used STRING. We identified 253 DEGs in the brown module, of which 242 were annotated for HUGO symbol and used for further functional analysis. The resulting network of 242 DEGs in the brown module had significantly more interactions than expected for a random set of proteins of similar size drawn from the genome (number of edges = 726, average node degree = 6, clustering coefficient = 0.438, PPI enrichment p value <1.0 × 10−16). Thirty-eight Reactome pathways were found to be significantly enriched for DEGs in the brown module. One enriched Reactome pathway, axon guidance (HSA-422475), was related to cytoskeleton and axon morphology (Table 3 and Figure 3).
Functionally enriched pathways associated with cytoskeleton and axon morphology from genes in the brown co-expression module in PIPN.

GO: gene ontology; PIPN: paclitaxel-induced peripheral neuropathy; KEGG: Kyoto Encyclopedia of Genes and Genomes; FDR: false discovery rate; Nbrown: count of genes in the brown module in the pathway; Npathway: count of genes in the pathway.

STRING connectivity network demonstrating a protein–protein interaction network of predicted functional partners for differentially expressed genes in the brown module. Nodes represent all proteins produced by a single protein coding gene locus. Edges represent specific or meaningful associations. Node color: axon guidance pathway (KEGG hsa04360) genes (red), second shell of interactors (white). Node size: protein of unknown three-dimensional (3D) structure (small), protein of known or predicted 3D structure (large). Color of the edges connecting the nodes represents the types of evidence supporting the connections: predicted gene neighborhood (green), predicted gene fusions (red), known interactions from experimental evidence (pink), co-expression (black), and text-mining (green). Disconnected nodes in the network are not displayed.
Discussion
This study is the first to provide molecular evidence that suggests that a number of cytoskeleton- and axon morphology-related mechanisms identified in various pre-clinical models of neuropathic pain25–27,59 are associated with chronic PIPN in breast cancer survivors. Of note, the regulation of actin cytoskeleton, focal adhesion, and axon guidance pathways was identified using both analytical methods.
The structural organization and dynamic remodeling of the neuronal cytoskeleton are responsible for cell migration and proliferation, as well as neuronal polarization and the establishment of a synaptic network. 60 In terms of the regulation of cytoskeleton and focal adhesion pathways, both are involved in intracellular signaling and in the regulation of cell motility. 61 Focal adhesions are large, dynamic protein complexes that enable the cytoskeleton to connect to the extracellular matrix of a cell. 62 At these specialized structures, integrin receptors cluster together to connect the extracellular matrix on the outside of the cell with the actin cytoskeleton within the cell. 63 These complexes transmit force or tension at adhesion sites to maintain strong attachments to the extracellular matrix, act as signaling centers for numerous intracellular pathways (e.g., processes involved in the reorganization of the actin cytoskeleton), modulate growth cones, and regulate axon regeneration of peripheral neurons.64,65 While no studies of PIPN were found, in a mouse model of oxaliplatin-induced peripheral neuropathy, 66 the absence of the advillin-containing focal adhesion protein (i.e., a sensory neuron specific actin binding protein) was associated with significant increases in cold allodynia. In addition, in a study of gene expression changes in patients with intractable neuropathic pain following spinal cord injury, 67 the focal adhesion pathway was one of the enriched pathways identified when these patients were compared to control patients without pain.
While originally identified as instructive cues to guide embryonic axons, axon guidance proteins have numerous functions including the control of synaptic plasticity. 68 Emerging evidence suggests that axon guidance mechanisms regulate the neuronal remodeling (e.g., dying back, axonal pruning) that occurs when peripheral nerves are injured.69,70 Consistent with our identification of an association between PIPN and perturbations and co-expression among genes in the axon guidance signaling pathway, in a recent pre-clinical model of PIPN, 71 the administration of paclitaxel acted directly on sensory neurons to alter inositol 1,4,5-triphosphate activity and initiated axonal degeneration. The authors concluded that paclitaxel-induced degradation differs from developmental degradation in the initiation of the degradation cascade and suggested that axon pruning molecules may be potential targets to prevent PIPN.
The hypoxia-inducible factor 1 (HIF-1) signaling pathway, a master regulator of cellular responses to hypoxia, 72 plays an important role in axon regeneration following peripheral nerve injury. 73 It has been found to mediate both processes that protect against axonal degeneration 74 and stimulate axon regeneration. 75 While no studies have reported on an association between PIPN and this pathway, findings from preclinical studies suggest that this transcription factor plays a significant role in the development of bortezomib-induced peripheral neuropathy, 76 diabetic peripheral neuropathy, 77 and sciatic nerve injury. 78
In our study, the co-expression of genes in the gap junctions signaling pathway was associated with PIPN. Gap junctions contain intracellular channels that facilitate cell-to-cell communications through direct exchange of intracellular messages. 79 The major gap junction proteins are from the connexin family (e.g., connexin 32). 80 Of note, connexin 32 is a fundamental protein in the peripheral nervous system that is associated with the x-linked form of Charcot-Marie-Tooth Disease, the second most common form of hereditary sensory and motor neuropathy. 81 While no studies were found using paclitaxel, in an ex vivo mouse sciatic nerve injury model, 82 prolonged exposure of the nerve to oxaliplatin caused a forced and persistent opening of connexin 32 channels and connexin 29 hemichannels in peripheral myelinated neurons which resulted in a disruption of axonal potassium homeostasis. This prolongation was almost completely blocked by the gap junction inhibitor octanol. The authors suggested that gap junction proteins may serve as neuroprotectants.
In terms of the enrichment of genes identified in the dendritic spine head GO annotated cellular component, dendritic spines are small, thin, specialized protrusions localized on excitatory synapses. They are primarily composed of polymerized actin or filamentous actin which undergo morphological changes in response to a stimulus. 83 While best characterized for their role in learning and memory, 84 emerging evidence suggests that the remodeling of dendritic spines may contribute to neuropathic pain by reorganizing nociceptive processing pathways 85 and abnormal dendritic spine structure following disease or injury may represent the “molecular memory” for maintaining chronic pain. 86 While no studies of CIPN were identified, findings from preclinical studies suggest that dendritic spine dysgenesis is involved in chronic pain associated with spinal cord injury,87,88 burns,89,90 and diabetes. 91 In terms of PIPN, because microtubules are thought to play a role in dendritic spine plasticity, 92 paclitaxel-induced stabilization of microtubules may adversely restrict morphological changes of dendritic spines and result in significant neurotoxicity.
Several limitations warrant consideration. While our sample size was relatively small, we have an extremely well-characterized sample of breast cancer survivors with and without PIPN. Future research with larger sample sizes may improve the resolution of the co-expression networks. Of note, no differences were found in the total cumulative dose of paclitaxel that the two groups of survivors received. Consistent with previous reports,93,94 we evaluated for differences in RNA expression from peripheral blood rather than from DRG neurons. Therefore, we can only infer that these findings are consistent with changes in the peripheral nervous system. Finally, our findings need to be replicated before they their translational impact can be explored. Part of the translational genomics 95 agenda to improve health 96 is to integrate high throughput molecular data with rich demographic and clinical data to evaluate mechanisms that underlie clinical conditions like PIPN. 97 Given the limits of pre-clinical models of pain,98,99 the identification of patterns of co-expression and perturbed pathways in survivors with PIPN that are related to mechanisms previously identified in pre-clinical models provides support for the translational value of this approach (e.g., findings such as these can help guide the development and selection of future pre-clinical models,98,99 or identify molecular features as targets 100 for pharmacological interventions 101 or as diagnostic biomarkers 102 ).
This study provides molecular evidence that a number of cytoskeleton- and axon morphology-related mechanisms identified in preclinical models of various types of neuropathic pain, including CIPN,19,103,104 are preferentially found in cancer survivors with persistent PIPN. The perturbations and co-expression patterns in these processes suggest persistent damage and/or changes in the regulation of the cytoskeleton and axon morphology in the peripheral nervous system of these survivors. Future studies need to evaluate for differences in epigenetic changes (i.e., methylation, microRNA) between survivors with and without PIPN, which may reflect changes in regulation patterns. In addition, studies are warranted that evaluate for common and distinct cytoskeleton- and axon morphology-related mechanisms associated with other neurotoxic CTX drugs (e.g., platinum, platinum and taxane combination).
Supplemental Material
MPX878088 Supplemental Material1 - Supplemental material for Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology in breast cancer survivors with chronic paclitaxel-induced peripheral neuropathy
Supplemental material, MPX878088 Supplemental Material1 for Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology in breast cancer survivors with chronic paclitaxel-induced peripheral neuropathy by Kord M Kober, Mark Schumacher, Yvette P Conley, Kimberly Topp, Melissa Mazor, Marilynn J Hammer, Steven M Paul, Jon D Levine and Christine Miaskowski in Molecular Pain
Supplemental Material
MPX878088 Supplemental Material2 - Supplemental material for Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology in breast cancer survivors with chronic paclitaxel-induced peripheral neuropathy
Supplemental material, MPX878088 Supplemental Material2 for Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology in breast cancer survivors with chronic paclitaxel-induced peripheral neuropathy by Kord M Kober, Mark Schumacher, Yvette P Conley, Kimberly Topp, Melissa Mazor, Marilynn J Hammer, Steven M Paul, Jon D Levine and Christine Miaskowski in Molecular Pain
Supplemental Material
MPX878088 Supplemental Material3 - Supplemental material for Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology in breast cancer survivors with chronic paclitaxel-induced peripheral neuropathy
Supplemental material, MPX878088 Supplemental Material3 for Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology in breast cancer survivors with chronic paclitaxel-induced peripheral neuropathy by Kord M Kober, Mark Schumacher, Yvette P Conley, Kimberly Topp, Melissa Mazor, Marilynn J Hammer, Steven M Paul, Jon D Levine and Christine Miaskowski in Molecular Pain
Supplemental Material
MPX878088 Supplemental Material4 - Supplemental material for Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology in breast cancer survivors with chronic paclitaxel-induced peripheral neuropathy
Supplemental material, MPX878088 Supplemental Material4 for Signaling pathways and gene co-expression modules associated with cytoskeleton and axon morphology in breast cancer survivors with chronic paclitaxel-induced peripheral neuropathy by Kord M Kober, Mark Schumacher, Yvette P Conley, Kimberly Topp, Melissa Mazor, Marilynn J Hammer, Steven M Paul, Jon D Levine and Christine Miaskowski in Molecular Pain
Footnotes
Acknowledgments
Recruitment was facilitated by Dr. Susan Love Research Foundation’s Army of Women® Program. Anatol Sucher managed the storage and processing of the biospecimens.
Declaration of Conflicting Interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Cancer Institute (NCI, CA151692) and the American Cancer Society (ACS, IRG-97–150-13). Dr Miaskowski is supported by grants from the ACS and NCI (CA168960). This project was also supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI Grant Number UL1 TR000004. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.