
Abstract
Protein kinase M ζ is well known for its role in maintaining memory and pain. Previously, we revealed that the activation of protein kinase M ζ in the anterior cingulate cortex plays a role in sustaining neuropathic pain. However, the mechanism by which protein kinase M ζ is expressed in the anterior cingulate cortex by peripheral nerve injury, and whether blocking of protein kinase M ζ using its inhibitor, zeta inhibitory peptide, produces analgesic effects in neuropathic pain maintained chronically after injury, have not previously been resolved. In this study, we show that protein kinase M ζ expression in the anterior cingulate cortex is enhanced by peripheral nerve injury in a transcription-independent manner. We also reveal that the inhibition of protein kinase M ζ through zeta inhibitory peptide treatment is enough to reduce mechanical allodynia responses in mice with one-month-old nerve injuries. However, the zeta inhibitory peptide treatment was only effective for a limited time.
Introduction
The question of how memory is permanently stored even though the physical traces of memory such as synaptic connections are not permanent had not been solved. An atypical protein kinase C (PKC) isoform, protein kinase M ζ (PKMζ), has recently emerged as the answer to this question.1–4 This kinase has the unique property that it lacks a regulatory subunit. Thus, PKMζ is constitutively active once it is expressed and this feature enables this kinase to maintain memory. 5 PKMζ is necessary and sufficient for maintaining long-term potentiation (LTP) and many kinds of memories. Blocking of PKMζ using zeta inhibitory peptide (ZIP) disrupts hippocampal LTP after the induction phase.1,6 Several types of memories such as fear, spatial, and taste aversion memories can be erased by ZIP treatment to the hippocampus, amygdala, and insular cortex.2,5,7,8 At the molecular level, PKMζ maintains memory by keeping GluA2-containing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) in the postsynaptic density (PSD) regions.9–11
Neuropathic pain induced by peripheral nerve injury can lead to LTP-like changes in the anterior cingulate cortex (ACC), a brain region known to be involved in affective dimension of pain.12–15 The mechanism of LTP maintenance is thought to be fairly universal in the brain. Based on this similarity, we previously hypothesized that PKMζ in the ACC could mediate the maintenance of neuropathic pain and showed that peripheral nerve injury enhances the PKMζ expression in the ACC.16,17 We also revealed that the inhibition of PKMζ in the ACC has an analgesic effect. Intriguingly, PKMζ in the ACC sustains GluA1-containing AMPAR in the PSD region. However, it has not been clearly elucidated whether ZIP is effective for treating neuropathic pain that is chronically maintained. It is also important to investigate how long the effects of ZIP continue after a single treatment.
In this study, we examined whether LTP or long-term depression (LTD) stimulation of the ACC leads to PKMζ activation. We also measured the level of PKMζ mRNA in the ACC after peripheral nerve injury. In addition, we investigated the effect of ZIP on neuropathic pain induced one month prior to treatment and evaluated the duration of the effect of single ZIP treatments to the ACC.
Materials and methods
Animals
Male wild-type C57BL/6NCrljBgi mice (6–10 weeks old) were purchased from Orient Bio. The mice were maintained in a 12-h light/dark cycle. Food and water were provided ad libitum. All experiments were conducted according to the guidance of the Institutional Animal Care and Use Committee of Seoul National University.
Cannula implantation and drug infusion
Guide cannulas (24 gauge) were implanted bilaterally into the ACCs of mice (+0.7 mm, ± 0.4 mm, and −1.7 mm) anesthetized with a ketamine/xylazine mixture. The mice were given at least one week to recover after cannula implantation. A 30-gauge injection cannula was then implanted 0.2 mm lower than the guide. For the intra-ACC infusion, 0.5 μl ZIP (10 nmol/μl) 16 or actinomycin D (ActD) (20 ng/μl), 18 or vehicle was delivered bilaterally within 1 min and the cannula remained for an additional 1 min after the drug microinfusion was completed. After all experiments were completed, the mice brains were processed to assess the injection site. Mice that were cannulated outside of the ACC were excluded from the analysis.
Neuropathic pain surgery
Mice were anesthetized with a ketamine/xylazine mixture (5.9:1) in saline. Their eyes were protected by artificial tear jelly or saline. The left leg of each mouse was shaven using scissors and sterilized with a 70% alcohol and povidone iodine liquid. About 1 cm of the left thigh skin was cut, exposing the muscles. An incision was made in the muscle using scissors, and sterile saline was applied to the exposed region. Next, the common peroneal nerve (CPN) was ligated with a wax-coated braided silk suture 4–0 without disturbing or including the blood vessel. The ligature was slowly tightened until twitching of the dorsiflexors of the foot became visible at the digit. After making a knot, the skin was sutured using a 5–0 silk suture and cleaned with povidone iodine liquid. In a few cases, the mice did not show any allodynia response three days after CPN ligation. These mice were therefore excluded from further experiments.
Measurement of mechanical allodynia response
Mechanical allodynia responses were measured essentially as described previously. 19 The mice were placed in individual cylinders and allowed to acclimatize for 1 h prior to testing. Mechanical allodynia was assessed based on the responsiveness of the hind paw to the application of a von Frey filament to the point of bending. Positive responses included licking, biting, and sudden withdrawal of the hind paw. Mechanical pressure from a 1.65 filament (force, 0.008g) was used to test the mice’s mechanical allodynia nine times with inter-trial intervals of 5 min. The animals were then permitted to rest for 2 h after drug infusion, and their mechanical allodynia was retested. All behavioral experiments were performed by a blind experimenter.
Subcellular fractionation for PSD fraction
The purification of PSD fraction was performed essentially as described previously. 20 The CPN-ligated mice were anesthetized and decapitated 2 h after ZIP infusion. Three slices (400 μm) of the ACC region near the infusion site were collected per mouse. Six of the ACC slices were used for fractionation. Briefly, the slices were homogenized in Frac buffer (30 mM pH 7.4 Tris-Cl, 4 mM EDTA, and 1 mM EGTA) containing a protease inhibitor cocktail. The homogenates were centrifuged at 500g, at 4°C for 5 min, twice, to remove the nucleus fraction and debris. The supernatants were centrifuged at 100,000g for 1 h at 4°C, and the pellet was lysed using Frac buffer containing 0.5% Triton X-100 and a protease inhibitor cocktail. After incubation for 20 min on ice, the lysates were carefully loaded onto the surface of 1 M sucrose and then centrifuged at 100,000g for 1 h at 4°C. The pellet (PSD fraction) was used for blotting after which it had been lysed using PSD lysis buffer (1 M pH 7.4 HEPES, 5 M NaCl, 10% Triton X-100, 10% sodium deoxycholate, 10% sodium dodecyl sulfate (SDS), and 100 mM DTT) containing the protease inhibitor cocktail.
Western blot analysis
Western blot was performed essentially as described previously.21,22 The mice were lightly anesthetized with isoflurane and then decapitated. The region of the ACC (400 μm thickness slice, three slices per mouse) was dissected and then homogenized in RIPA buffer (50 mM pH 7.6 Tris-Cl, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 1 mM DTT, 0.5% sodium deoxycholate) containing a protease inhibitor cocktail and protein phosphatase inhibitor cocktail after glycine treatment (30 min) and washout (60 min). After centrifugation, the supernatants were used for protein quantification by Bradford assay. Electrophoresis of equal amounts of total protein was performed on 4%–12% SDS-polyacrylamide gels (Invitrogen). The separated proteins were transferred onto a nitrocellulose membrane and stored at 4°C overnight. After blocking with 3% bovine serum albumin (for PKMζ and p-PKMζ) or 5% skim milk (for actin) in Tris-buffered saline plus Triton X-100 for 2 h at room temperature, the membranes were incubated with PKCζ (1:500, Invitrogen), phosphor-PKCζ (1:1000, Cell Signaling), GluA2 (1:1000, Abcam), or actin (1:5000, Sigma) primary antibody at 4°C overnight. After washing, the membranes were treated with a horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature, followed by enhanced chemiluminescence detection of the proteins according to the manufacturer’s instructions. The density of immunoblots was measured and analyzed using ChemiDoc™ MP System (Bio-Rad).
Quantitative real-time PCR
Quantitative real-time PCR was performed essentially as described previously. 23 To measure mRNA expression level in the ACC after nerve injury, the region of the ACC (400 μm thickness slice, three slices per mouse) was dissected. Total RNA was purified with Trizol (Invitrogen) or RNAiso plus (Takara) reagent according to the user’s manual. After DNase I treatment for 15 min at room temperature, purified RNA was used for cDNA synthesis prepared by the SuperScript ® III First-Strand Synthesis System for RT-PCR (Cat. #18080–051, Invitrogen). After phenol/chloroform extraction and ethanol precipitation, cDNA was used for quantitative real-time PCR. To compare PKMζ mRNA levels between the sham and nerve injury groups, quantitative real-time PCR was performed using SYBR Premix Ex Taq II (Cat. #RR820A, Takara) in a CFX96 Real-Time PCR Detection System according to the user’s manual. The primers for PKMζ are 5′-ACGCCCACCTTCGGTAGAGC-3′ for forward and 5′-GGACGTGGCAGCGTTTATGG-3′ for reverse. The primers for brain-derived neurotrophic factor (BDNF) are 5′-AGTGTAATCCCATGGGTTACACCA-3′ for forward and 5′-CAGGAAGTGTCT ATCCTTATGAATCG-3′ for reverse. The primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) are 5′-TGCACCACCAACTGCTTA-3′ for forward and 5′-GGATGCAGGGATGATGTTC-3′ for reverse. The expression level of PKMζ or BDNF was normalized to the expression level of GAPDH as a reference gene.
LTP recording using MED64
Mice were anesthetized with isoflurane and killed by decapitation. The brain was removed and then quickly placed in ice-cold, oxygenated (95% O2, 5% CO2) cutting solution containing (in mM) 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 MgSO4, 15 Glucose, and 2 CaCl2. Coronal slices (300 μm) of the ACC were prepared using a vibratome (Leica VT 1000S). Those slices were allowed to recover in oxygenated artificial cerebrospinal fluid (aCSF) at 26°C for at least 2 h before recordings were performed. aCSF contained the following (in mM): 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 MgSO4, 15 Glucose, and 2 CaCl2. Multielectrode array system (Panasonic, MED64) was used to record extracellular field excitatory postsynaptic potential
Data analysis
Statistical comparisons were made using unpaired or paired t-tests or one-way ANOVAs. All data were presented as the mean ± SEM. In all cases, p < 0.05 was considered as statistically significant.
Results
It has been reported that LTP stimulation enhances PKMζ and p-PKMζ levels in hippocampal slices. 25 Thus, we tested whether LTP stimulation also activates PKMζ in the ACC. To induce chemical LTP, ACC slices were treated with 1 mM glycine for 30 min and incubated for 1 h after being rinsed. Consistent with previous findings, 26 this protocol induced LTP for 1 h after the glycine washout (Figure 1(a)). The p-PKMζ expression was enhanced by chemical LTP stimulation, although PKMζ level showed a tendency to increase (Figure 1(b)). However, the PKMζ mRNA level was significantly reduced after glycine treatment (Figure 1(c)). Next, we tested whether LTD also affects PKMζ activation in the ACC. However, mGluR-dependent LTD stimulation induced by several doses of dihydroxyphenylglycine bath-application for 30 min did not affect the level of PKMζ and p-PKMζ in the ACC (Figure 1(d)). Given that peripheral nerve injury such as amputation can induce an LTP-like state in the ACC, these results imply that nerve injury induces LTP and it may, then, enhance and activate PKMζ in the ACC in a transcription-independent manner.

We previously showed that cAMP signaling increased PKMζ expression within a short period of time. 16 This result indicates a transcription-independent expression of PKMζ, because transcription of PKMζ pre-mRNA is likely to take more than 30 min.25,27 To clarify this point, we tested the change of PKMζ mRNA in the ACC three days after nerve injury. As expected, the PKMζ mRNA level did not change significantly in the nerve injury group (Figure 1(e), upper panel). Quantitative real-time PCR also showed no increase in PKMζ mRNA level in the ACC after nerve injury (Figure 1(e), lower panel). Moreover, treatment with the transcription inhibitor ActD did not block the increase in PKMζ protein level in the ACC four days after nerve injury (Figure 1(f)). The absence of an effect of ActD on PKMζ protein level was not due to the use of a low dose of ActD (10 ng/side, bilateral infusion), as this dose is sufficient to block the increase in BDNF mRNA level in the ACC of mice injected with formalin to induce acute inflammatory pain (Figure 1(g)). 28 These results further support the idea that the increase in PKMζ level in the ACC induced by nerve injury was independent of transcription.
Inhibition of PKMζ using ZIP reduces allodynia responses shown in neuropathic pain. 16 In the case of neuropathic pain, noxious signals from the peripheries are continuously delivered to various brain areas including the ACC region. Thus, it is useful to determine how long the alleviating effects of a single ZIP treatment for hyperalgesia last. Based on our previous result, 16 we measured the mechanical allodynia response at 4 or 6 h after ZIP infusion into the ACC. As shown in Figure 2(a) and (b), there was still a noticeable analgesic effect after an interval of 4 h, but not at 6 h. As neuropathic pain is a type of chronic pain, it is necessary to evaluate the effect of ZIP on chronically maintained neuropathic pain in view of clinical trial. In a previous study, the effects of ZIP were tested in a neuropathic pain model that was 3 or 7 days old. 16 Therefore, we tested the effects of ZIP on mice with nerve injuries induced one month before and found that ZIP still reduced the allodynia response in nerve-injured mice after one month (Figure 2(c) and (d)).
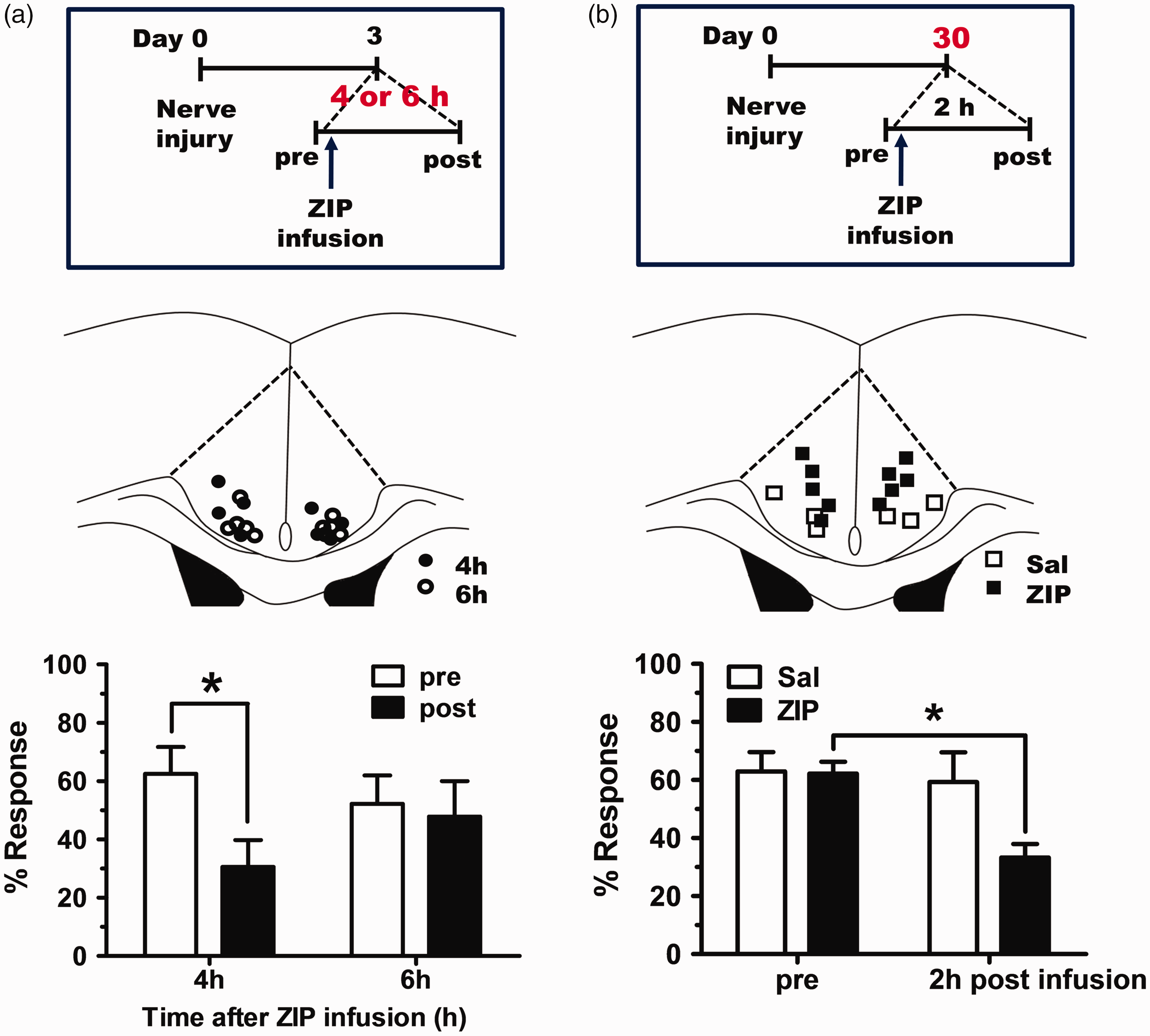
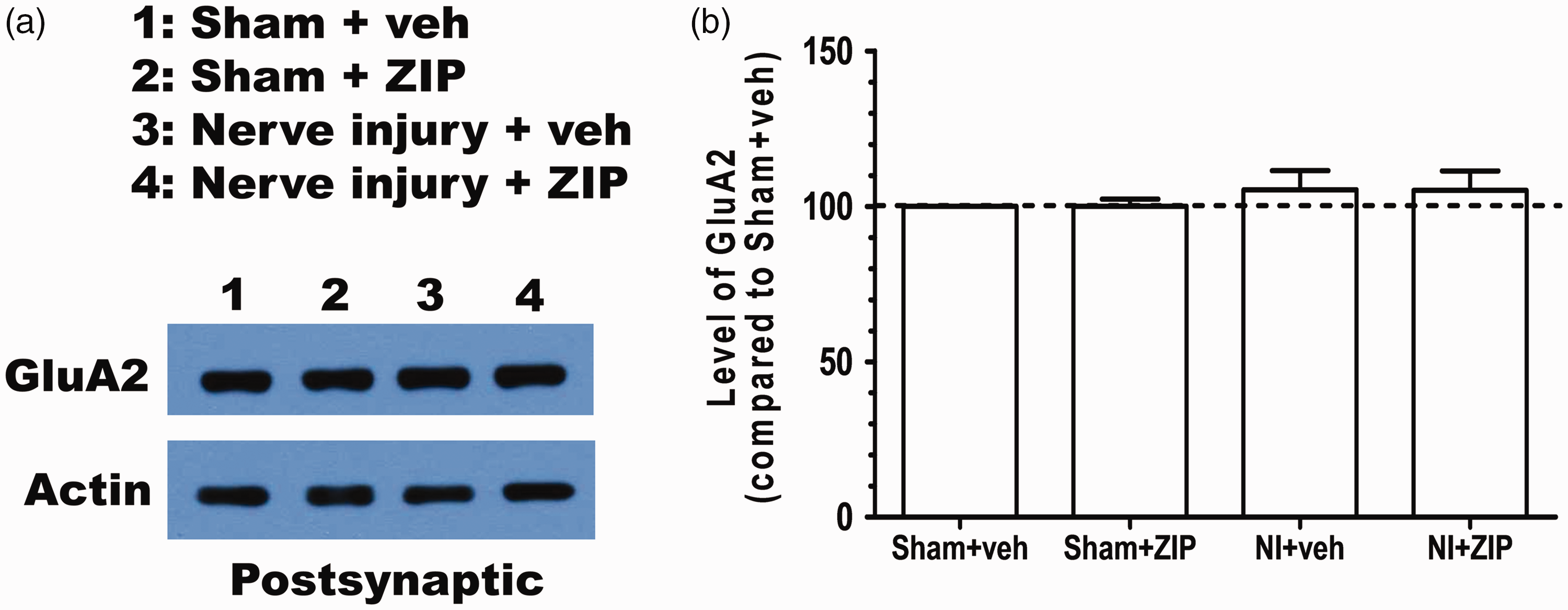
Our results shed some light on the synaptic mechanisms likely to be responsible for the analgesic effects produced by ZIP in neuropathic pain. PKMζ can postsynaptically potentiate the amplitude of AMPA receptor-mediated excitatory postsynaptic currents. 29 Given that glutamatergic synaptic transmission in the ACC is increased after nerve injury, 30 PKMζ may contribute to the maintenance of the enhanced synaptic transmission induced by nerve injury. Previously, we showed that ZIP infusion in the ACC reduced postsynaptic GluA1, one component of AMPARs, selectively in the nerve injury group. 16 This result is surprising because previous studies have identified GluA2 as a target of PKMζ.9,10 Thus, we tested if GluA2 is also reduced by ZIP treatment in the ACC. However, there was no difference in GluA2 between the saline and ZIP-infused groups in nerve-injured mice (Figure 3). This result indicates that PKMζ exerts its effect through the GluA1 AMPAR subunit at synapses in the ACC.
Discussion
Here, we revealed that LTP, not LTD, stimulation activates PKMζ in the ACC. These enhancements in PKMζ and p-PKMζ are independent of transcription. The level of PKMζ mRNA in the ACC did not show enhancement after peripheral nerve injury. In addition, we confirmed that ZIP is still effective against mechanical allodynia maintained chronically in mice with neuropathic pain but only within a limited time. Although we did not observe the increase in PKMζ mRNA in the ACC three days after nerve injury, it does not mean that the expression of PKMζ is completely independent of transcription. It is plausible that PKMζ mRNA increases during a short time period after the initial peripheral nerve injury. Ongoing local cortical activity after nerve injury can activate adaptive mechanisms in the neurons of the ACC. Thus, to reveal the exact mechanism of PKMζ enhancement induced by nerve injury, it is necessary to measure the PKMζ mRNA level a relatively short time after nerve injury.
Most patients suffering from neuropathic pain have been afflicted for long time, rather than a short period. If ZIP is to be used as a medicine, it should show therapeutic effect in chronically maintained neuropathic pain. ZIP infusion into the ACC still reduces the mechanical allodynia response shown in neuropathic pain a month old (Figure 2(b)). Given that ZIP application into the ACC does not show any side effects such as memory deficits, 16 this finding raises the possibility that ZIP could be used as a medication for chronic pain. However, the therapeutic effect of ZIP did not last for more than 4 h (Figure 2(a)). Therefore, it is necessary to apply ZIP continuously via a drug delivery system or alternatively to develop long-lasting inhibitor against PKMζ.
Given that PKMζ regulates GluA2 subunit trafficking in the synapse, it is quite interesting that the inhibition of PKMζ did not reduce GluA2 in the PSD fraction of nerve-injured mice. This discrepancy might stem from brain region- or modality-specific functions of PKMζ. To date, GluA2 has been found to be a target of PKMζ in the hippocampus. However, we found that GluA1 is a target of PKMζ in the ACC. It is possible that PKMζ acts through different targets depending on brain regions. Another possibility is that PKMζ works with different targets depending on neurophysiological functions. Although we tested a target of PKMζ in the ACC of nerve-injured mice, if we examined a target of PKMζ in the ACC of fear-conditioned mice, GluA2 might be a target of PKMζ. Regardless of which hypothesis is true, at least it is certain that PKMζ has a direct downstream target other than N-ethylmaleimide-sensitive fusion (NSF) protein to regulate the trafficking of GluA1 because NSF does not directly bind to GluA1. 31 Our previous finding that the GluA1, and not the GluA2, subunit is required for LTP in the ACC supports our present results because LTP in the ACC underlies chronic pain. 32 Thus, it is valuable to investigate which target molecule of PKMζ in the ACC mediates GluA1 trafficking in the synapse to sustain chronic pain. These future studies will contribute to the development of new medicines for chronic pain.
Footnotes
Author Contributions
H-GK designed the study, carried out all the experiments, and outlined and wrote the manuscript. SY carried out the molecular experiments. D-HH, PP, and C-SL performed LTP recordings using MED64. KL, MZ, and B-KK supervised the experiments, participated in the interpretation of the data, and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We are grateful to Hyunjun Jung and Yoonkey Nam at Department of Bio and Brain Engineering, Korea Advanced Institute of Science and Technology for the technical help with MED64 probes.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by two National Research Foundation (NRF) of Korea grants funded by the Korean government (MSIP) [NRF-2012R1A3A1050385 to B-KK and 2018R1C1B6008530 to H-GK]. SY was supported by the BK21 Research Fellowship from the Ministry of Education, Science and Technology, Republic of Korea.