
Abstract
Reward system has been proved to be important to nociceptive behavior, and the nucleus accumbens (NAc) is a key node in reward circuitry. It has been further revealed that dopamine system modulates the NAc to influence the pain sensation, whereas the role of glutamatergic projection in the NAc in the modulation of chronic pain is still elusive. In this study, we used a complete Freund’s adjuvant-induced chronic inflammatory pain model to explore the changes of the glutamatergic terminals in the NAc, and we found that following the chronic inflammation, the protein level of vesicular glutamate transporter1 (VGLUT1) was significantly decreased in the NAc. Immunofluorescence staining further showed a reduced expression of VGLUT1-positive terminals in the dopamine receptor 2 (D2R) spiny projection neurons of NAc after chronic inflammatory pain. Furthermore, using a whole-cell recording in double transgenic mice, in which dopamine receptor 1- and D2R-expressing neurons can be visualized, we found that the frequency of spontaneous excitatory postsynaptic currents was significantly decreased and paired-pulse ratio of evoked excitatory postsynaptic currents was increased in D2R neurons, but not in dopamine receptor 1 neurons in NAc of complete Freund’s adjuvant group. Moreover, the abnormal expression of soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex contributed to the reduced formation of glutamate vesicles. Hence, our results demonstrated that decreased glutamate release in the indirect pathway of the NAc may be a critical mechanism for chronic pain and provided a novel evidence for the presynaptic mechanisms in chronic pain regulation.
Keywords
Introduction
Chronic pain is a complicated symptom which involves multiple information processes such as somatosensation, emotion, and reward. Reward circuits have long been hypothesized to be important to the representation of pain. 1 Evidences from preclinical models and human patients suggested that the mesolimbic reward circuitry is critical for pain sensation and pain-related emotional experiences.2,3 The nucleus accumbens (NAc) is a key node in reward circuitry, which integrates the mesencephalic dopaminergic signals, 4 glutamatergic information from ventral hippocampus (vHC), frontal cortex, and amygdala. 5 The important role of this convergence in NAc also has been revealed in both acute and chronic pain states from human imaging studies. 6
Accumulated animal studies have investigated that the input and output information in NAc modulates nociceptive and pain-related affective behaviors.7,8 Arising activation of NAc-projecting DA neurons in the ventral tegmental area (VTA) can suppress tonic pain, 9 and releasing of endogenous opioids and substance P from the midbrain could also inhibit the nociception.10,11 Pioneering works have proven that VTA-NAc circuitry modulates thermal hypersensitivity using optogenetics methods and have provided evidence that brain-derived neurotrophic factor protein presented an antinociceptive role. 8 As to the presynaptic afferent input of NAc, the complex interplay of glutamate, dopamine, and other neuropeptides modulates the synaptic function and neuronal excitability of the NAc synchronously. However, as the major excitatory neurotransmitters in the NAc, the changes of glutamatergic input information under chronic pain condition still need further elucidation.
The release of glutamate in the presynaptic area depends upon the expression and the function of secretory vesicles, vesicular glutamate transporters (VGLUTs). VGLUT family presents distinct expression patterns. 12 VGLUT1 and VGLUT2 are the major secretory vesicles in the brain, and VGLUT3 often acts as a cotransporter of glutamate and other neurotransmitters, such as serotonin, gamma-aminobutyric acid (GABA), and acetylcholine. 12 Among the distinct functions of VGLUTs, changes of VGLUT1 and VGLUT2 expression can represent the glutamatergic input conditions of the NAc. 13 Presynaptic glutamate release is also closely related to the vesicle fusion when it depends on the vesicle synthesis, and soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) complex can regulate the release rhythm of presynaptic neurotransmitters by regulating the homeostasis of vesicle synthesis and fusion. 14 However, the role of SNARE in regulating glutamatergic synaptic homeostasis has not been well-characterized under chronic pain condition.
Outputs from the NAc are organized into two primary projection pathways – direct pathway and indirect pathway, which can be distinct with dopamine receptor 1 (D1R) and dopamine receptor 2 (D2R). These two parallel, opposing pathways could charge with affective evaluation of salient events that shape behaviors.15,16 Dopamine serves as opposite effects on these two pathways. Dopamine could enhance the activity of D1R-expressing spiny projection neurons (SPNs) but suppress that of D2R-expressing SPNs, which indicates that identical presynaptic information could lead to elicit diversified responses from different circuits in the NAc. 17 However, the critical roles of glutamatergic afferents into these two pathways responding to the inflammatory pain remain largely unknown.
In the present study, we examined the expression pattern and quantified the protein expression levels of VGLUT1 and VGLUT2 in the NAc using a well-established chronic inflammatory pain model. In addition, we compared the different glutamatergic afferent changes of the two distinct circuits under a pain condition to reveal the potential remodeling in NAc circuitry. Furthermore, we explored the SNARE complex alteration after pain stimulus to provide evidence of the presynaptic changes in neurotransmitter release. The present results demonstrated that pathway-specific changes resulting from glutamatergic presynaptic releasing alteration led to a disruption of circuit balance in NAc, which may contribute to the comorbidity of chronic pain and anxiety.
Materials and methods
Experimental animals and pain model
The generation of D1R-tdTomato and D2R-enhanced green fluorescent protein (eGFP) transgenic mice has been previously described. 18 All experiments were conducted in adult male C57BL/6 mice weighing 20 to 22 g. Adult mice were housed in a temperature-controlled environment on a 12-h light/dark cycle with free access to food and water. Animal procedures were in compliance with the Institutional Animal Care and Use Committee at the Fourth Military Medical University (Xi’an, China) and carried out according to the “Principles of Medical Laboratory Animal Care” issued by the National Institutes of Health (NIH). All efforts were made to reduce animal suffering and decrease the number of animals used. The complete Freund’s adjuvant (CFA)-induced inflammation pain model was established as previously described.20 Mice were injected with 10 μL of CFA (1mg/mL Mycobacterium tuberculosis; Sigma, UK) into the plantar surface of the right hind paw to induce inflammation under isoflurane anesthesia. The mice for electrophysiological recording and Western blot were injected with same amount of CFA into both hind paws.
Behavioral tests
All behavioral tests were performed in awake, unrestrained mice in an appropriately lighted, quiet room. All the tests were carried on in habituated mice by an observer blinded to the identity of the groups.
Nociceptive behavioral test
The von Frey test was performed as previously described. 19 Mechanical sensitivity was tested with manual application of von Frey hairs (North Coast, USA). The plantar surface of hind paw was selected as the stimulating part. Each filament was applied 10 times, and the paw withdrawal response was recorded. The proper force would make the von Frey hairs bended 90° rapidly. Hargreave’s test was tested by application of radiant heat light to the plantar surface of hind paw. The response latency was measured by an automated readout (IITC Life Science, USA).
Open field test
The open field test (OFT) was performed at day 8 after the injection as previously described. 20 The test was carried out as previously described. 21 Mice were placed in the center of a cubic chamber (40 × 40 × 40 cm). Behaviors were observed for 10 min under a dim light, and the travel trace was measured by an automated analysis system (SMART 3.0, Panlab S.L.U., Spain). After each test, the arena was cleaned with 75% alcohol solution. Four measures of behavior were recorded: the total distances, the time in the central area, the entries into the central area, and the distances in the central area.
Marbles burying test
This test was processed after OFT. First, the cage was prepared with wood chip bedding approximately 5 cm deep, lightly tamped down to make a flat surface. Mice were put into the cage for habituation for 20 min. Then, 12 regular glass marbles were put on the surface, each about 4 cm apart, evenly spaced. Then, one mouse was placed in the cage, and a video camera was used to record for 20 min. When the entire test was finished, the video was replayed to analyze the number of marbles buried.
Elevated plus maze test
This test was performed after the marbles burying test, using an elevated plus maze (EPM) apparatus and elevated 50 cm above the floor. The EPM was constructed of black Plexiglas, consisting of two opposing open arms (50 × 10 cm), two opposing closed arms (50 × 10 ×40 cm), and a central area (10 × 10 cm). Behaviors were observed for 5 min under a dim light, and the travel trace was measured by SMART 3.0. After each test, the arena was cleaned with 75% alcohol solution. Four measures of behavior were scored: the total distances, the time in the open arms, the entries into the open arms, and the distance in the open arms.
Western blot
After the behavioral tests, mice were anesthetized with isoflurane, and tissues of the NAc were collected by using the 1-mm stainless steel brain matrix (68707, RWD, China) to locate the frontal plane (anterior/posterior: from +1.70 to +0.98 mm) and separating it on the iced culture dish immediately, according to The Mouse Brain in Stereotaxic Coordinates (second edition, George Paxinos and Keith B. J. Franklin) and put into radioimmunoprecipitation assay lysis buffer (10 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.5% NP-40, and 1 mM ethylenediaminetetraacetic acid at pH 7.4) quickly consisting protease and phosphatase inhibitors (Roche, CH). Lysates were cleared by centrifugation (12,000 r/min for 30 min). Protein concentration was measured using the BCA Protein Assay Kit (Thermo, USA) and metered all of the samples into 2 μg/μL. The samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to 0.22 μm polyvinylidene difluoride membranes (Millipore, USA). After 2 h in slim milk at room temperature, the membranes were immunoblotted with primary antibodies at 4° overnight. Then, membranes were incubated with horseradish peroxidase-conjugated anti-rabbit or mouse IgG antibody (1:1000, Cell Signaling Technology, USA) for 1 h at room temperature. The primary antibodies were as follows: rabbit IgG against Syntaxin 1A (STX1A, 1:1000, Cell Signaling Technology, USA), synaptosome-associated protein-25 (SNAP-25, 1:1000), vesicle-associated membrane protein 2 (VAMP2, 1:1000), mammalian uncoordinated 18–1 (Munc18–1, 1:1000), NSF (1:1000), VGLUT2(1:1000), mouse IgG against VGLUT1 (1:500, Millipore, USA), and mouse IgG against glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1000, Zhuang Zhi, China). The images were scanned by Chemiluminescent Imaging System (5200Multi, Tanon, China) and quantified by using Image J software (Version 1.48). GAPDH served as the standard.
Immunofluorescence staining
For immunofluorescence studies, the mice were deeply anesthetized with isoflurane and then transcardially perfused with 30 mL of 0.01M phosphate-buffered saline (PBS; pH 7.4), followed by 50 mL of 4% paraformaldehyde (PFA) in phosphate buffer (PB, pH 7.4). The brains were removed and fixed in 4% PFA for 2 h, then put into 30% sucrose at 4°C for 24 h. The whole brains were cut into 30-μm-thick sections on a cryotome (Thermo, USA). The sections were blocked with 5% goat serum in PBS containing 0.3% Triton X-100 for 4 h at room temperature. The sections were incubated at 4°C for two days with a mixture of anti-VGLUT1 rabbit antibody (1:200, Synaptic Systems, Germany) or anti-VGLUT2 guinea pig antibody (1:200, Synaptic Systems, Germany) and mouse IgG anti-microtubule-associated protein 2 (MAP2) (1:500, Abcam, UK) or anti-Synaptophysin 1 (SYN1, 1:500, Synaptic Systems, Germany) in 0.01 M PBS containing 0.3% Triton X-100, 0.25% λ-carrageenan, and 1% donkey serum. The sections were further incubated for 2 h at room temperature with a mixture of Alexa 488 or Alexa 594-conjugated donkey anti-mouse or rabbit or guinea pig IgG (1:800, Invitrogen, USA). To differentiate VGLUT1 and VGLUT2 colocalized with D1 or D2 SPNs, the brain sections from D1R-tdTomato and D2R-eGFP double transgenic mice were used for the staining. The sections were incubated at 4°C for two days with a mixture of both anti-VGLUT1 rabbit antibody (1:200, Synaptic Systems, Germany) and anti-VGLUT2 guinea pig antibody (1:200) in 0.01 M PBS containing 0.3% Triton X-100, 0.25% λ-carrageenan, and 1% donkey serum. The sections were further incubated for 2 h at room temperature with a mixture of Alexa 488 (VGLUT1/D1R) or Alexa 594 (VGLUT1/D2R)-conjugated donkey antirabbit (1:800, Invitrogen, USA) and Alexa 647 (VGLUT2 with D1R or D2R)-conjugated donkey anti-guinea pig IgG (1:800, Jackson ImmunoResearch, USA). Images were captured using a laser scanning confocal microscope (FV1000, Olympus, Japan). The density of VGLUT1- or VGLUT2-positive terminals, the colocalization of VGLUT1 or VGLUT2 terminals and SYN1, the contacts of VGLUT1 or VGLUT2 terminals with MAP2, and the number of colocalization of VGLUT1, VGLUT2 terminals and D1R or D2R were all measured by using Image-Pro Plus (Version 6.0).
In vitro electrophysiology recording
The slice preparations were followed by previously described. 22 Briefly, mice were anesthetized with pentobarbital sodium (30–40 mg/kg body weight) and transcardially perfused with 20 mL of ice-cold carbogenated (95% O2, 5% CO2) cutting solution containing (in mM): 115 choline-chloride, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 8 MgCl2, 26 NaHCO3, 10 D-(+)-glucose, 0.1 L-ascorbic acid, and 0.4 sodium pyruvate (with osmolarity of 300–305 mOsm/l). The brains were then rapidly removed and placed in ice-cold cutting solution for slice preparation. The coronal slices (300 µm) containing the NAc were prepared by a slicer (VT1200s, Leica, Germany) and then incubated in a holding chamber at 32°C with carbogenated cutting solution for 15 to 20 min. The slices were then transferred to artificial cerebral spinal fluid containing (mM): 119 NaCl, 2.3 KCl, 1.0 NaH2PO4, 26 NaHCO3, 11 D-(+)-glucose, 1.3 MgCl2, 2.5 CaCl2 (pH 7.4, with osmolarity of 295–300 mOsm/l) at room temperature for at least 1 h. Whole-cell patch clamp recordings were performed with infrared differential interference contrast visualization at 25°C to 28°C. Recording pipettes were filled with a solution as previously described. 22 The tdTomato-positive neurons or eGFP-positive neurons in the NAc were held at −70 mV. The signals were not obtained until 3 min after establishing whole-cell mode. Cells with series resistance more than 20 MΩ at any time during the recordings were discarded. Neurons with resting membrane potentials more negative than −60 mV, and action potentials with overshoot were selected for further experiments. For the spontaneous excitatory postsynaptic currents (sEPSCs) results, 3-min duration recordings were performed in gap-free voltage-clamp mode. Last 2 min sEPSCs events were automatically detected and manually checked again with MiniAnalysis (6.0.3, Synaptosoft, USA). For paired-pulse ratio (PPR) experiments, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-mediated EPSCs were evoked by a concentric bipolar electrode (CBARC75, FHC, USA) placed in the caudal edge of the core in the NAc. A stimulus pulse lasted for 200 μs, and the interstimulus interval of two stimulus pulse was 50 ms. The PPR was calculated with the peak current response to the second pulse divided by that of the first response. Recordings of evoked EPSCs (eEPSCs) and sEPSCs were performed in the presence of picrotoxin (100 μM) and AP-5 (50 μM) to block activation of GABAA receptors and N-methyl-D-aspartate receptors respectively. The recordings were obtained using a multiclamp 700B or axopatch 200B amplifier (Molecular Devices, USA) filtered at 5 kHz and sampled at 20 kHz with a Digidata 1550B (Molecular Devices, USA) to store on a computer. Clampex 9.0 (Molecular Devices, USA) was used for data acquisition and off-line analysis.
Statistical analysis
All data are presented as the means ± the standard error of mean, and p < 0.05 was considered significant. All statistical analysis and graph plotting were performed using GraphPad Prism 7.0 (GraphPad Software, Inc., USA). Statistical comparison of two means was performed by unpaired two-tailed Student’s t test, while multiple comparisons were performed using the two-way ANOVA analysis of variance followed by the Bonferroni’s post hoc test. And n indicates the number of animals tested unless indicated otherwise.
Results
CFA-induced chronic pain model presented hyperalgesia and anxiety behaviors
To explore the potential changes of the NAc under chronic pain conditions, we used a well-established CFA-induced chronic inflammatory pain model (Figure 1(a) and (b)). Before CFA injection, we measured the baseline of paw withdrawal latencies (PWL) in response to radiant heat stimulation and paw withdrawal thresholds (PWT) in response to von Frey hairs, and no significant difference between the CFA group and saline group was found. Intense thermal hyperalgesia and mechanical allodynia stably occurred on the day 3 and the day 2 after CFA injection, respectively, and persisted over one week (Figure 1(a) and (b); day 3, Hargreave’s test, p = 0.0332; day 2, von Frey test, p = 0.029). Compared with the baseline, there was a dramatic increase of mechanical response frequency seven days after CFA injection, but this was not observed in saline group (Figure 1(c) and (d); saline group, p > 0.05; CFA group, p < 0.01). These data suggested that CFA could induce a robust and persistent mechanical and thermal hyperalgesia.

CFA-induced chronic pain model presented thermal, mechanical hyperalgesia and a comorbidity of anxiety. (a) Time course showed the CFA increased both thermal pain hypersensitivity (b) and mechanical pain hypersensitivity gradually. (c) After the injection for seven days, the mechanical pain hypersensitivity of saline group had no change (d) but of CFA group increased significantly. (e) Representative motion trials of the saline group and CFA group in the OFT. (e to i) The CFA group presented significant anxiety behavior compared with the saline group in OFT. (f) Total distance analysis in OFT showed that CFA had no effect on total distance. The entries (g), time (h), and distance (i) in central area showed a significant reduction comparing with the saline group. (j) Representative motion trials of the saline group and CFA group in the EPM test. (j to n) The CFA group presented significant anxiety behavior compared with the saline group in EPM test. (k) Total distance analysis in EPM test showed that CFA had no effect on total distance. The entries (l), time (m), and distance (n) in open arms showed that the CFA group presented a significant reduction comparing with the saline group. (o) Representative situation of marbles burying test between saline group and CFA group. (p) Time course showed the numbers of marbles buried per 5 min. (q) Total numbers of marbles buried after 20 min. The data are presented as the means ± SEM (n = 6/group; *p < 0.05, **p < 0.01, ***p < 0.001, compared to the saline group).
Then, we asked whether the chronic pain model also presented anxiety behaviors. To address this issue, we performed three kinds of paradigms to measure the anxiety behaviors of mice. Initially, we conducted an OFT, and there was no significant difference in total distance between CFA group and saline group (Figure 1(e) and (f); p = 0.6327), suggesting the motor function was not influenced by the inflammatory pain. Then, we compared the entries in the central area, time in central area, and distance traveled in central area, which were indicators of anxiety behaviors. We found a significant decrease in CFA group comparing with saline group in entries, time, or distance in the central area (Figure 1(g) to (i); entries, p = 0.04; time, p = 0.02; distance, p = 0.024). Similarly, in EPM test, no significant difference in total distance was found (Figure 1(j) and (k); p = 0.20), and CFA group presented a decrease in the entries, time, and distance in the open arms (Figure 1 (l) to (n); entries, p = 0.016; time, p = 0.04; distance, p = 0.015). As to the marble burying test, we counted the numbers of marbles buried both in CFA group and saline group within 20 min. Mice in CFA group presented much more marbles remaining on the surface of padding (Figure 1(o) and (q); p < 0.001), and the number of marbles buried was relatively less compared with saline group in a time-dependent manner (Figure 1(p); p < 0.001 at 5 min and p < 0.01 at 10 min). Above results indicated that CFA-induced chronic pain caused evident comorbid anxiety behaviors in mice.
Chronic inflammatory pain decreased VGLUT1 expression in the presynaptic terminals of the NAc
To determine the glutamatergic and GABAergic presynaptic changes in the NAc under chronic pain condition, we performed Western blot to assess the total protein levels of VGLUT1, VGLUT2, and Vesicular GABA Transporter (VGAT). CFA injection induced a robust downregulation of VGLUT1 protein in the NAc (Figure 2(a) and (b); p = 0.037), but not VGLUT2 and VGAT (Figure 2(a) , (c) and (d); VGLUT2, p = 0.123; VGAT, p=0.521). Then, to further investigate whether the downregulation of VGLUT1 occurred in the presynaptic areas, we performed the double immunohistochemistry to observe the density of collocated terminals of VGLUT1, VGLUT2, and SYN1, which is the major synaptic vesicle protein. Double immunofluorescence showed that the VGLUT1 and SYN1 were well-coexisted in axon terminals in the NAc (Figure 3(A)), and the immunostaining density of VGLUT1–positive terminals was decreased in CFA group compared with saline group (Figure 3(C); p = 0.042), similarly as the density of VGLUT1/SYN1-colocalized terminals (Figure 3(D); p = 0.02). The VGLUT2-immunoreactive terminals were also positive for SYN1 (Figure 3(B)); however, there were no significant changes in the density of both VGLUT2-immunopositive terminals and VGLUT2/SYN1 dual positive terminals (Figure 3(E) and (F); p = 0.846, p = 0.842, respectively).

CFA-induced chronic pain caused a selective decrease in VGLUT1 expression in the NAc. (a) Western blot showed the protein levels of VGLUT1, VGLUT2, and VGAT. (b to d) Quantitative analysis revealed the expression changes of VGLUT1, VGLUT2, and VGAT in CFA group comparing with saline group. The data are presented as the means ± SEM (n = 5/group *p < 0.05, compared to the saline group).

The presynaptic expressions of VGLUT1 in the NAc were decreased. (A) Double immunostaining for VGLUT1 (red, (a) and (b)) and SYN1 (green, (c) and (d)) in axon terminals in the NAc. (e) and (f) are merged figures of (a) and (c), and of (b) and (d), respectively. Insets are enlarged image of dashed areas in each figure. Arrowheads indicate VGLUT1 and SYN1-colocalized terminals. (B) Double immunostaining for VGLUT2 (red, (a) and (b)) and SYN1 (green, (c) and (d)) in axon terminals in the NAc. (e) and (f) are merged figures of (a) and (c), and of (b) and (d), respectively. Insets are enlarged image of dashed areas in each figure. Arrowheads indicate VGLUT2 and SYN1-colocalized terminals. (C and D) Quantitative analysis revealed that the immunostaining density of VGLUT1-positive terminals and VGLUT1/SYN1 coexisted terminals was decreased in the CFA group compared with saline group. (E and F) Quantitative analysis revealed that the immunostaining density of VGLUT2-positive and VGLUT2/SYN1-double-positive terminals was unaltered in the CFA group compared with saline group. Scale bar = 10 µm. The data are presented as the means ± SEM (n = 5/group *p < 0.05, compared to the saline group).
Then, we asked whether the decreased presynaptic terminals presented less contact with neurons in the NAc. We performed double immunohistochemistry for VGLUT1, VGLUT2, and MAP2, which serves to regulate the structure and stability of microtubules in axons and dendrites. Similarly, the density of VGLUT1-immunopositive axon terminals was observed to decrease significantly (Figure 4(A) and (C); p = 0.0198), and the VGLUT1/MAP2-colocalized terminals also presented reduction (Figure 4(A) and (D); p = 0.0462). No significant difference was found in density of VGLUT2-positive terminals and VGLUT2/MAP2 double stained terminals between CFA group and saline group (Figure 4(B), (E), and (F); p = 0.66, p = 0.76, respectively).

The contact of VGLUT1-positive terminals with neurons of the NAc was decreased. (A) Double immunostaining for VGLUT1 (red, (a) and (b)) and MAP2 (green, (c) and (d)) in the NAc. (e) and (f) are merged figures of (a) and (c), and of (b) and (d), respectively. Insets are enlarged image of dashed areas in each figure. Arrowheads indicate VGLUT1-positive terminals contacting with MAP2-positive structure. (B) Double immunostaining for VGLUT2 (red, (a) and (b)) and MAP2 (green, (c) and (d)) in the NAc. (e) and (f) are merged figures of (a) and (c), and of (b) and (d), respectively. Insets are enlarged image of dashed areas in each figure. Arrowheads indicate VGLUT2-positive terminals contacting with MAP2-positive structure. (C and D) Quantitative analysis revealed that the immunostaining density of VGLUT1-positive terminals and the contact between VGLUT1-positive terminals and MAP2-positive neuronal processes were decreased in the CFA group compared with saline group. (E and F) Quantitative analysis revealed that the immunostaining density of VGLUT2-positive terminals and the contact between VGLUT2 and MAP2-positive structures were unaltered in the CFA group compared with saline group. Scale bar = 10 µm. The data are presented as the means ± SEM (n = 5/group *p < 0.05, compared to the saline group).
Chronic inflammatory pain impaired glutamatergic synaptic transmission in the indirect pathway of the NAc
To further clarify the cell types in the NAc which influenced by the chronic pain stimuli, we adopted a well-designed transgenic mice line. This D1R-tdTomato and D2R-eGFP double transgenic mice could visualize the neurons located in the direct pathway and indirect pathway as previously described. 18 We then performed whole-cell patch clamp in the tdTomato- or the eGFP-positive neurons (Figure 5(a) and (b)), and these neurons presented a typical firing pattern of SPNs, which yield a slow ramp depolarization and delay to the initial spike with the depolarization current step stimulation (Figure 5(c) and (d)). Next, using the double transgenic mice, we conducted CFA-induced pain model, and recording the sEPSCs in the SPNs expressing D1R and D2R. We found a significant decrease in the frequency of sEPSCs when recording the D2R-expressing neurons, but not for the amplitude (Figure 5(g) and (h); frequency, p = 0.02; amplitude, p = 0.94). However, as to the D1R SPNs, we found no evident changes in both frequency and amplitude (Figure 5(e) and (f); frequency, p = 0.84; amplitude, p = 0.17). Then, we measured PPR (50 ms of interpulse interval) to further assess the presynaptic release probability in the SPNs expressing D1R and D2R. A significant enhancement in PPR was found in D2R SPNs, but not in D1R SPNs recordings in CFA group (Figure 5(i) and (j), D1R SPNs PPR, p = 0.768; D2R SPNs PPR, p = 0.0062). Next, we performed double immunohistochemistry of VGLUT1 and VGLUT2 with D1R-tdTomato and D2R-eGFP double transgenic mice. The number of VGLUT1/D2R double-stained terminals was also decreased compared with VGLUT1/D1R double-stained terminals in CFA group (Figure 6(A) (c), (d), and (f); p = 0.04), and it had no significant changes in saline group (Figure 6(A) (a), (b), and (e); p = 0.56). In addition, there were no significant changes in the numbers of VGLUT2/D1R-colocalized terminals when compared with VGLUT2/D2R in saline group (Figure 6(B) (a), (b), and (e); p = 0.47) or CFA group (Figure 6(B) (c), (d), and (f); p = 0.64). Above results indicated that the SPNs located in the indirect pathway received a weakened glutamate presynaptic modulation but that in the direct pathway was unaffected.

Differential effects of CFA-induced chronic pain on glutamatergic synaptic transmission in D1R- and D2R-expressing SPN. (a) A Drd1-tdTomato-positive neuron (D1R SPN) was recorded by patch clamp pipette in differential interference contrast (DIC) image mode. Scale bar = 20 μm. (b) A Drd2-eGFP-positive neuron (D2R SPN) was recorded by patch clamp pipette in DIC image mode. Scale bar = 20 μm. (c) The same neuron from (a) showed a slow ramp depolarization and delay to the initial spike in current clamp mode. (d) The same neuron from (b) showed the typical delay firing pattern in current clamp mode. (e and g) Representative recording traces of sEPSCs of D1R SPNs (E) and D2R SPNs (g). (f and h) Summary data of sEPSCs frequency and peak amplitude in D1R SPNs (f: D1 saline group, n = 9; D1 CFA, n = 5) and D2R SPNs (h: D2 saline, n = 6; D2 CFA, n = 6). (i) Representative traces (left) and summary data (right) of paired-pulse ratio (PPR) of D1R SPNs from saline group and CFA group (D1R SPNs PPR: saline n = 6, CFA n = 8, p = 0.768). (j) Representative traces (left) and summary data (right) of PPR of D2R SPNs from saline group and CFA group (D2R SPNs PPR: saline n = 7, CFA n = 9, p = 0.0062). The data are presented as the means ± SEM, *p < 0.05, **p < 0.01, compared to the saline group.

The density of VGLUT1-positive terminals was decreased in D2R neurons in CFA group. (A) Double immunostaining for D1R (red, (a) and (c)) and VGLUT1 (green, (a) and (c)), D2R (green, (b) and (d)) and VGLUT1 (red, (b) and (d)). Insets are enlarged image of dashed areas in each figure. Arrowheads indicate VGLUT1-positive terminals contacting with D1R or D2R neurons. Quantitative analysis revealed that the density VGLUT1-positive terminals contacting with D2R neurons was decreased compared with D1R neurons in CFA group (f), and it was unaltered in saline group (e). (B) Double immunostaining for D1R (red, (a) and (c)) and VGLUT2 (orange, (a) and (c)), D2R (green, (b) and (d)) and VGLUT2 (orange, (b) and (d)). Insets are enlarged image of dashed areas in each figure. Arrowheads indicate VGLUT2-positive terminals contacting with D1R or D2R neurons. Quantitative analysis revealed that the density of VGLUT2-positive terminals contacting with D1R and D2R had no significant difference both in saline group (e) and in CFA group (f). Scale bar = 10 µm. The data are presented as the means ± SEM (n = 5/group; *p < 0.05, compared with D1R in CFA group).
Abnormal SNARE complex contributed to the decreased release of presynaptic glutamate vesicles
Given the significant changes in the glutamatergic terminals in the NAc, the underlying mechanisms are still elusive. Then, we used Western blot to test the expression of SNARE complex which controls the vesicle formation and recycling. Comparing with the saline group, the protein levels of STX1A, SNAP-25, and NSF presented a relatively sharp reduction in the CFA group (Figure 7; STX1A, p = 0.027; SAP-25, p = 0.0012; NSF, p = 0.0015). And the expression of VAMP2 and Munc18–1 in the CFA group showed a similar expression level comparing with the saline group (Figure 7; VAMP2, p = 0.65; Munc18–1, p = 0.41). Above results illustrated that the abnormal expression of SNARE complex might contribute to chronic pain-induced reduction of presynaptic glutamate release.

Downregulation of SNARE correlative proteins in the NAc contributed to the impairment of glutamatergic synaptic transmission. (a) Western blot showed the protein expression of STX1A, SNAP-25, VAMP2, Munc18–1, and NSF. (b) Quantitative analysis of STX1A, SNAP-25, VAMP2, Munc18–1, and NSF comparing with GAPDH, the expression of VAMP2 and Munc18–1 had no change while that of STX1A, SNAP-25, and NSF was decreased in CFA group. The data are presented as the means ± SEM (n = 5/group; *p < 0.05, **p < 0.01 compared to the saline group).
Discussion
In this study, we found that CFA-induced chronic inflammatory pain decreased the presynaptic glutamate release via affecting the SNARE complex-mediated secretory vesicles recycling in the presynaptic membranes. Moreover, the following reduction VGLUT1 mainly influenced the inputs of D2R-expressing SPN, which led to an unbalanced status between direct pathway and indirect pathway in the NAc. These findings suggest that the nociceptive stimuli could weaken the excitatory glutamatergic information to the indirect pathway within the reward circuits, which may be intermingled with the dopaminergic modulation rhythmically.
The NAc receives glutamatergic projections from the basolateral nucleus of amygdala, 23 the vHC, 24 the prefrontal cortex (PFC), 25 and the paraventricular nucleus of the thalamus (PVT), 26 among which the PFC is the main VGLUT1 and PVT is the main VGLUT2 output sources. The VGLUT1 expressing PFC-NAc projection pathway modulates the activity of the limbic system and enables the behavioural flexibility, which provides top-down control of the sensory and affective process. Accumulating studies reported that PFC could regulate pain process,27,28 and there are also evidences proving that optogenetic activation of PFC-NAc projection could produce strong antinociceptive and anxiolytic effects in a neuropathic pain model, which provide an insight into corticostriatal glutamatergic system in pain regulation. 29 In our study, we also found that the expression of VGLUT1 was sharply decreased after the chronic inflammatory stimuli, which may also verify the critical role of glutamatergic synapses in the midbrain reward circuits. It will be interesting and important to differentiate the exact role of VGLUT1 projection to either D1 SPNs or D2 SPNs in the inflammatory pain model with optogenetics or chemogenetics methods in the future. For the VGLUT2, we did not find the significant difference between saline and CFA group, which only suggested that thalamostriatal synapses may be relatively normal in chronic inflammatory pain condition. However, we cannot exclude the possibility that thalamic activity could influence striatal state in other pain models. We also did not find the VGAT protein expression difference between saline and CFA group. But the inhibitory transmission of NAc is more complicate than the excitatory transmission since there are too many inhibitory presynaptic terminals intermingling in the NAc.30–32 To dissect out the changes from different inhibitory synapses of NAc in chronic inflammatory pain, further experiments need to be done with either optogenetics or chemogenetics combined with the different interneuron tools, such as specific promotors-driven Cre mouse lines or transgenetic mouse line with fluorescent reporter.
As to the mechanisms underlying the presynaptic changes, we found a series of alteration of SNARE complex under the chronic pain condition in the present study. STX1A is located at the presynaptic plasma membrane. It is a SNARE protein which is essential for synaptic vesicle fusion. 33 STX1A interacts with Munc 18–1, the molecule that modulates SNARE complex formation in return. 34 As to SNAP-25, which is on neuronal presynaptic membranes, forms a core complex with STX1A and VAMP2 to mediate synaptic vesicle fusion with the plasma membrane during Ca2+-dependent exocytosis. 35 And VAMP2 is predominantly inserted in presynaptic vesicle membranes, 36 assembly of which with the STX1A and SNAP-25 is a critical event necessary for vesicle fusion and neurotransmitter release. NSF is a hexameric ATPase and then associates with the α-SNAP/SNARE complex to mediate SNARE disassembly during membrane fusion. The ATPase activity of NSF induces eventual recycling of the SNARE complex for subsequent membrane fusion. 37 In our study, we found a significantly decreased protein level of STX1A, SNAP-25, and NSF which are all important factors for the vesicle formation and recycling, and it suggests that the chronic nociceptive stimuli would induce an abnormal of presynaptic vesicle system; thus, the presynaptic information would be hard to recruit enough secretary vesicles to release glutamate, leading to a reduced glutamatergic release (Figure 8). In addition, D2R-class receptors also expressed at presynaptic site and could decrease cortical inputs.38–40 Therefore, more experiments are necessary to carry out to confirm the related mechanisms and find out other potential related events happened in the process of glutamate releases under chronic pain.
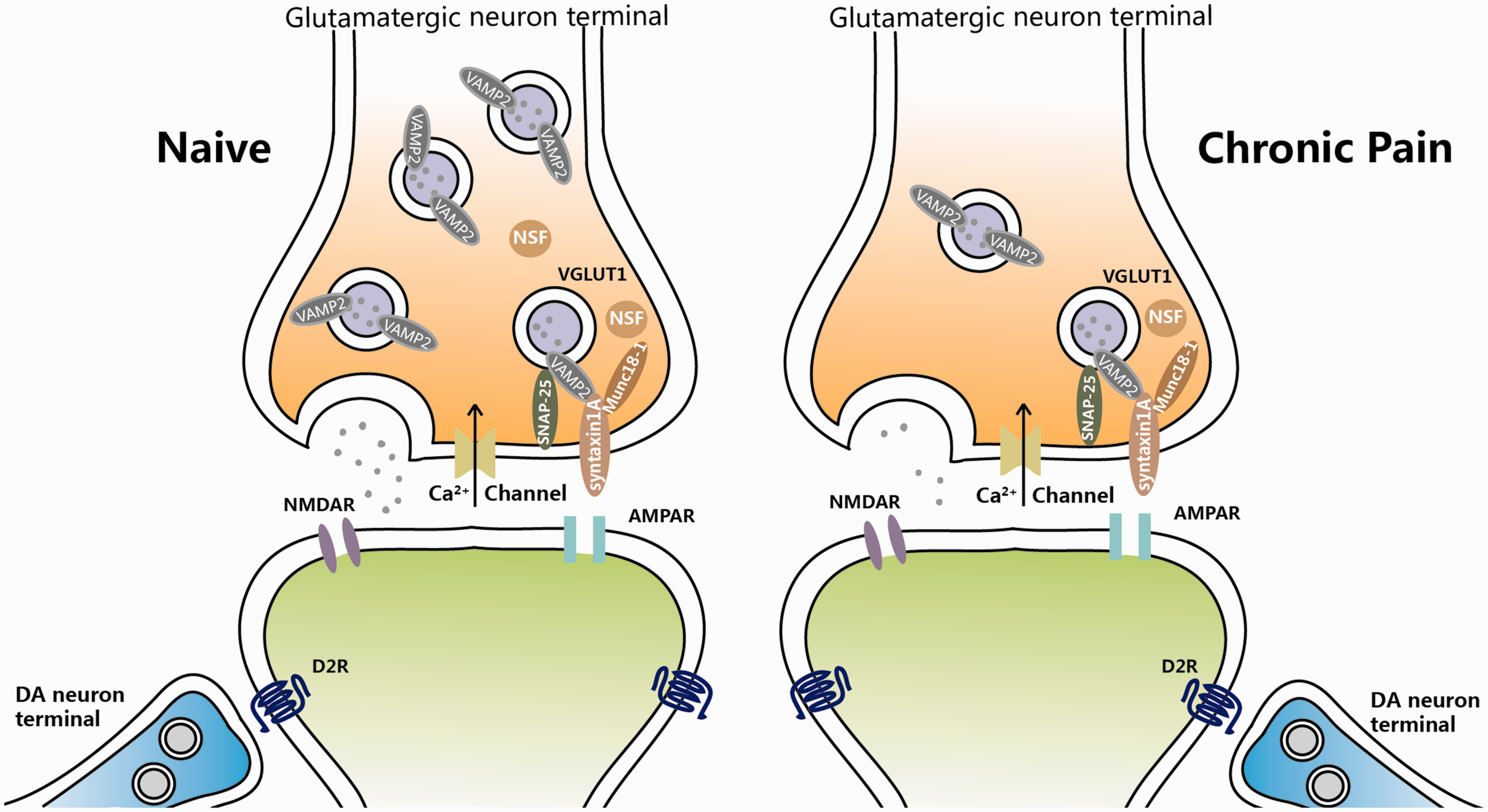
Schematic illustration of probable mechanisms for presynaptic glutamatergic terminal changes in the NAc driving chronic pain. See Discussion section for details.
In addition to the presynaptic mechanisms, previous studies have also shown that glutamate receptors in the NAc can regulate pain sensitivity.41,42 Persistent neuropathic pain has been found to selectively increase GluA1 subunit levels at the synapse in the NAc,42,43 the process of which seems to be trafficking increasing. And selective increase in GluA1 levels at synapses leads to the formation of Ca2+-permeable GluA2-lacking AMPAR, which could modulate the synaptic strength, via regulating the induction 39 of long-term potentiation or long-term depression.42,44 In this work, we found the reduction of glutamate signaling in presynaptic release, which may be the preorder events to the increased glutamate receptor functions, and seems to be certain compensation in the NAc under the chronic pain. The SPNs in the NAc are involved in two distinct pathways, with a balanced regulation pattern. Previous study has found that the peripheral nerve injury could cause a cell-specific upregulation in the indirect pathways of the NAc, which worsened the tactile allodynia. 7 However, how the synaptic connectivity was influenced by the presynaptic glutamate signaling was not clarified. And in this study, we confirmed that it was the abnormal presynaptic vesicles’ formation of the indirect pathways that causes the imbalance within the NAc, which may affect the control of the segmental withdrawal circuitry mediated by indirect pathway.
In the present study, there are several conclusions from the results. First, we found the glutamatergic terminals in the NAc under chronic pain condition released fewer glutamates, which was caused by the decreased expression of VGLUT1, but not VGLUT2. Second, our results suggested that the SPNs in the indirect pathway were vulnerable to the weakened glutamatergic input especially. Finally, the novel changes of glutamatergic terminals were due to the abnormal SNARE-mediated vesicle formation and recycling following CFA-induced chronic pain. These results shed new light on the specific neural circuits remodeling involved in chronic pain and its comorbidity and also provide a further evidence for the therapeutic approach targeting the reward circuits system in the brain.
Footnotes
Acknowledgments
The authors thank Dr. Guoping Feng for sharing the D1R-tdTomato transgenic mice and Dr. Charles Gerfen for sharing the D2R-eGFP transgenic mice. The authors also thank Dr. Yilin Wu for the assistance on the drawing of schematic illustration.
Author Contributions
WW and SW conceived the project and designed the experiments. CQ, BG, and KR performed the experiments; CQ, BG, HY, MW, TS, HL, and RL analyzed the data. WW and SW prepared the manuscript based upon the draft by BG and CQ. HL and RL revised it critically for important intellectual content. WW and SW provided the financial and administrative support for this project. All authors approved the final version of the manuscript submitted for publication, all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (81730035 and 81371240 to SW, 81771476 to WW); Innovation Teams in Priority Areas Accredited by the Ministry of Science and Technology (No. 2014RA4029 to SW); and Shaanxi Key Research and Development Program (2017SF-137 to WW).