
Abstract
Treating bone cancer pain continues to be a clinical challenge and underlying mechanisms of bone cancer pain remain elusive. Here, we reported that sonic hedgehog signaling plays a critical role in the development of bone cancer pain. Tibia bone cavity tumor cell implantation produces bone cancer-related mechanical allodynia, thermal hyperalgesia, and spontaneous and movement-evoked pain behaviors. Production and persistence of these pain behaviors are well correlated with tumor cell implantation-induced up-regulation and activation of sonic hedgehog signaling in primary sensory neurons and spinal cord. Spinal administration of sonic hedgehog signaling inhibitor cyclopamine prevents and reverses the induction and persistence of bone cancer pain without affecting normal pain sensitivity. Inhibiting sonic hedgehog signaling activation with cyclopamine, in vivo or in vitro, greatly suppresses tumor cell implantation-induced increase of intracellular Ca2+ and hyperexcitability of the sensory neurons and also the activation of GluN2B receptor and the subsequent Ca2+-dependent signals CaMKII and CREB in dorsal root ganglion and the spinal cord. These findings show a critical mechanism underlying the pathogenesis of bone cancer pain and suggest that targeting sonic hedgehog signaling may be an effective approach for treating bone cancer pain.
Introduction
Bone cancer pain is one of the most common complications in bone cancer patients. Bone cancer pain is severe and intractable and affects approximately 60% of patients with primary or metastatic bone cancer. 1 Despite decades of studies and numerous implicated processes, the specific cellular and molecular mechanisms that underlie the pathogenesis of bone cancer pain remain unclear, and the clinical approaches for its treatment are very limited.
Recently, we found that some of the proteins that play an important role in development are re-activated and play critical roles in bone cancer pain, such as EphB 2 and Wnt.3,4 Here, we found that sonic hedgehog (Shh) may be another such protein. Shh is a secreted glycoprotein that plays a crucial role in central nervous system development during embryogenesis. 5 Shh signaling is a highly conserved pathway among vertebrates and invertebrates. The components of Shh signaling in vertebrates primarily include the Shh ligand, the patched (Ptch) and smoothened (Smo) receptors, and Gli transcription factors.5,6 In the canonical pathway, activation of Shh signaling via the binding of Shh to Ptch results in the activation of Smo and the nuclear translocation of Gli.7,8 Emerging findings suggest that Shh plays important roles in the formation of neuronal circuits and synaptic plasticity.5,6 Shh could increase both the size of the presynaptic terminals and the frequency of miniature excitatory postsynaptic currents at the hippocampal neuron synapses.9,10 Recently, it was reported that Shh signaling was involved in nociceptive regulation.11–13 Shh mutation resulted in a lack of nociceptive sensitization in drosophila. 11 In the present study, we investigated the role of Shh signaling in the pathophysiology of bone cancer pain. We hypothesized that Shh signaling plays a critical role in bone cancer pain. Spinal blocking of Shh signaling inhibits the induction and maintenance of bone cancer pain. The possible mechanisms by which Shh signaling regulates bone cancer pain include regulating the excitability of dorsal root ganglion (DRG) neurons by modulating intracellular Ca2+, the activation of the GluN2B receptor, and subsequent Ca2+-dependent signals.
Materials and methods
Animals, anesthesia, drugs, and administration
Adult female Sprague-Dawley rats (160–180 g) (Xuzhou Medical University Animal Center, Xuzhou, China) were used. This study was carried out in strict accordance with the recommendation in the guide for the care and use of laboratory animals of the national institutes of health. The protocol was approved by the Committee on the Ethics of Animal Experiments of Xuzhou Medical University. The rats were housed in a temperature-controlled environment with 12-hr light-dark cycles and were fed standard laboratory diet and water ad libitum. All animals were handled daily for five days before the start of the experiment to minimize the stress reaction to manipulation. All surgeries were done under anesthesia with sodium pentobarbital (50 mg/kg, i.p.), and all efforts were made to minimize suffering. For in vivo test, cyclopamine (10 µg/10 µl) (Selleck Chemicals), Shh signaling inhibitor, were administrated intrathecally by lumbar puncture. For in vitro test, Shh-IgG (50 ng/ml or 500 ng/ml) (Sigma) and cyclopamine (5 µM) were delivered by bath application. The doses we chose were according to previous studies. 14
Model of bone cancer pain
Tumor cells were extracted from ascetic fluid of rats that received Walker 256 mammary gland carcinoma cells. Tumor cell implantation (TCI) was mimicked by injecting the tumor cells (1 × 105 cells/µl, 5 µl) into the intramedullary space of the right tibia to induce bone cancer in rats. In brief, following induction of general anesthesia with sodium pentobarbital (50 mg/kg, intraperitoneally), the animal was placed abdominal side up. A 1 cm rostro-caudal incision was made in the skin overlying the top half of the tibia after disinfecting with 75% v/v ethanol. Tumor cells were then injected into the intramedullary space of the right tibia. The injection site was closed using bone wax while the syringe was removed. The wound was dusted with penicillin powder and then closed. Rats in the sham group were injected with the same volume of boiled cells. The protocol was similar to that previously described in literature. 15
Behavioral test
Mechanical allodynia was determined by measuring incidence of foot withdrawal in response to mechanical indentation of the plantar surface of each hindpaw with a sharp, cylindrical probe with a uniform tip diameter of approximately 0.2 mm provided by an Electro Von Frey (ALMEMO 2390–5 Anesthesiometer, IITC Life Science Inc., Woodland Hills, CA). The probe was applied to six designated loci distributed over the plantar surface of the foot. The minimal force (in grams) that induced paw withdrawal was read off the display. Threshold of mechanical withdrawal in each animal was calculated by averaging the six readings, and the force was converted into millinewtons (mN).
Thermal hyperalgesia was determined by significant shortened latency of foot withdrawal in response to heat stimulation (IITC Model 336 Analgesia Meter, Series 8; IITC Life Science Inc.). In brief, the heat source was focused on a portion of the hind paw, and a radiant thermal stimulus was delivered to that site. The stimulus shut off automatically when hind paw moved (or after 20 s to prevent tissue damage). Thermal stimuli were delivered three times to each hind paw at 5- to 8-min intervals.
Spontaneous and movement-evoked pain-like behaviors were also analyzed. Spontaneous nociceptive behaviors were evaluated by measuring spontaneous guarding and flinching over a 2-min period of observations. Movement-evoked pain was assessed by measuring the time spent guarding over a 2-min period of observations after non-nociceptive palpation, and the limb use during spontaneous ambulation, which was scored on a scale of 0 to 4: 0 = normal use, 1 = slightly limping, 2 = clearly limping, 3 = no use of the limbs (partial), and 4 = no use of the limbs (complete).
Choronic constriction injury
To produce peripheral nerve injury, choronic constriction injury (CCI) model was established. Under anesthesia, the left common sciatic nerve of each rat was exposed at the mid-thigh level. Proximal to the sciatic nerve’s trifurcation was freed of adhering tissue and four ligatures (4–0 surgical catgut) were tied loosely around it with approximately 1 mm between ligatures. Animals in the sham group received surgery identical to that described in CCI but without nerve ligation.
Western blotting
To identify temporal expression of Shh signaling (Shh, Ptch1, Smo, Gli1), whole-cell protein extract lysates were used. To identify the activation of Shh signaling, nuclear extracts were prepared using an NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce Biotechnology) according to the manufacturer’s instruction. L4-L5 spinal cord segments were quickly removed from deeply anesthetized rats and homogenized in ice-cold RIPA lysis buffer containing a cocktail of protease inhibitors. Total proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to 0.2 µm polyvinylidene difluoride membrane. The following primary antibodies were used: anti-Shh (1:2000, Abcam), anti-Ptch1(1:1000, Sigma), anti-Smo (1:2000, Abcam), anti-Gli1 (1:2000, Abcam), anti-p-GluN2B (Tyr1472) (1:500, Millipore), anti-p-CaMKII (1:1000, Cell signaling Tech), anti-p-CREB (1:1000, Cell signaling Tech), anti-Histone H3 (1:1000, Abcam), and anti-GAPDH (1:10,000, Sigma). The filters were developed using ECL reagents (Perkin Elmer) with secondary antibodies from Millipore Bioscience Research Reagents. Data were analyzed with a Molecular Imager (Gel DocTM XR, 170–8170) and the associated software Quantity One-4.6.5 (Bio-Rad Laboratories).
ELISA assay
The ipsilateral sciatic nerves were rapidly remove and homogenized in ice-cold 0.01 mol/L phosphate-buffered saline. Protein concentrations were determined by bicinchoninic acid protein assay. Besides, the ipsilateral tibia lavage fluids were collected. The tibia was lavaged five times with 5 ml of ice-cold phosphate-buffered saline. The recovered fluid was centrifuged at 1000 r/min for 5 min at 4°C, and the supernatant was collected for further test. All tissues were collected from deeply anesthetized rats. The level of Shh concentration was measured by enzyme-linked immunosorbent assay using Shh N-Terminus Quantikine ELISA Kit (R&D systems), according to the manufacturer’s instruction. 16
Excised, intact DRG preparation
DRG neurons in excised ganglia that were prepared as described previously in literature 17 were tested in place. The isolated DRG neurons were from a mixed population of cells from the sciatic nerve, not just those that innervate the tibia. Briefly, the sciatic nerve was transected at midthigh level, and its proximal portion traced to the ganglia. A laminectomy was performed. Ice-cold, oxygenated, buffered solution containing (in mmol/L) 140 NaCl, 3.5 KCl, 1.5 CaCl2, 1MgCl2, 4.5 HEPES, 5.5 HEPES-Na, and 10 glucose (pH 7.3; osmolarity 310–320 mmol/L) was dripped onto surface of ganglion during procedure. The ganglia from TCI side of L4 and L5 segments were removed and placed in 35-mm petri dishes containing ice-cold, oxygenated, buffered solution. The perineurium was peeled off, and attached sciatic nerve and dorsal roots were transected at site adjacent to the ganglion. Intact ganglion was then treated with collagenase (type P, 1 mg/ml; Roche Diagnostics) for 30 min at 35°C and then incubated at room temperature for patch-clamp recordings.
Whole-cell current-clamp recordings
To test excitability of the nociceptive DRG neurons, whole-cell current-clamp and voltage-clamp recordings were made with an Axopatch-200B amplifier (Molecular Devices, Union city, CA) in the small DRG neurons (soma diameter: 15–30 µm) from the intact ganglion preparations. These neurons largely correspond to neurons with C-fiber conduction velocities. Conduction velocity was not measured in this study. The protocols were similar to that we have previously described. Glass electrodes were fabricated with a Flaming/Brown micropipette puller (P-97, Sutter instruments, Novato, CA). Electrode impedance was 3 to 5 MΩ when filled with saline containing (in mM) 120 K+-gluconate, 20 KCl, 1 CaCl2, 2 MgCl2, 11 ethylene glycol-bis-(β-aminoethyl-ether) N,N,N′,N′,-tetraacetic acid, 2 Mg-ATP, and 10 HEPES-K (pH 7.2, osmolarity 290–300 mOsm). Electrode position was controlled by a three-dimensional hydraulic micromanipulator (MHW-3, Narishige). When the electrode tip touched the cell membrane, gentle suction was applied to form a tight seal (serial resistance 2 GΩ). Under −70 mV command voltage, additional suction was applied to rupture the cell membrane. After obtaining the whole-cell mode, the recording was switched to current-clamping mode, and the resting membrane potential (RMP) was recorded.
All the DRG cells accepted for analysis had an RMP of −45 mV or more negative. To compare the excitability of the DRG neurons, we examined the RMP, action potential current threshold (APCT), and repetitive discharges evoked by a standardized intracellular depolarizing current. The RMP was taken 2 to 3 min after a stable recording was first obtained. APCT was defined as the minimum current required evoking an action potential by delivering intracellular currents from −0.1 to 0.7 nA (50 ms pulses) in increments of 0.05 nA. The whole-cell input capacitance (Cin) was calculated by integration of the capacity transient evoked by a 10 mV pulse in voltage clamp mode. Repetitive discharges were measured by counting the spikes evoked by, 1000 ms, intracellular pulses of depolarizing current normalized to 2.5 times APCT. All electrophysiological recordings and data analyses were conducted by experimenters blind to previous treatment of the cells.
Measurement of intracellular Ca2+ ([Ca2+]i)
Intact DRG were prepared and incubated in artificial cerebro-spinal fluid containing Fura-2/AM (5 µM) and Pluronic F-127 (0.5 mg/ml) (Invitrogen, Inc.). Fluorescence in the small- and medium-sized DRG neurons (diameter, 15–45 µm), but not the glial cells surrounded, was measured at 340 nm and 380 nm excitation and 520 nm emission (Olympus IX51 with ORCA-R2 digital camera, Hamamatsu Inc., Japan). The 340/380 nm emission ratio was used to determine [Ca2+]i. After each recording, 4-bromo A-23187 (BR-A, 10 µM, Sigma) was used to check the viability of the cells.
Data analysis
SPSS Version 15 (SPSS Inc., Chicago, IL) was used to conduct all the statistical analysis. The behavioral responses to mechanical and thermal stimuli over time among groups were tested with two-way analysis of variance with repeated measures followed by Bonferroni post hoc tests. Western blot data between groups were analyzed using nonparametric Kruskal–Wallis test, followed by Mann–Whitney U tests. The normality was tested by Shapiro–Wilk test (W test). One-way analysis of variance or t test was used to analyze intracellular Ca2+ activity and Shh concentration. Chi-squared tests were used to identify differences in the incidence of effects. All data are presented as mean ± SEM. The criterion for statistical significance was P < 0.05.
Results
Hedgehog signaling up-regulated and activated in nociceptive pathway during bone cancer pain
Bone cancer-induced changes in DRG and the spinal cord are critical for bone cancer pain generation. We began by investigating the protein expression of hedgehog signaling. Western blot analysis showed that TCI produced a significant increase in the expression of Shh, Ptch1, Smo, and Gli1 in DRG and the spinal cord, respectively. In DRG, Shh increased at postoperative day 3 and peaked at days 14 to 21. Ptch1, Smo, and Gli1 increased at postoperative day 5 and peaked at day 14 (Figure 1(a)). In the spinal cord, Shh increased at postoperative day 5; the other proteins for hedgehog signaling increased at day 7 and were maintained at a high level from day 7 to day 21 until the last examination (Figure 1(b)). However, the expression levels of hedgehog signaling proteins in the sham group were very low in both DRG and the spinal cord (Figure 1(a) and (b)). Gli1 is a nuclear transcription factor that will transfer to the nucleus with hedgehog signaling activation.7,8 To prove the activation of Shh signaling, the expression of Gli1 in nuclear extract was investigated. The expression of Gli1 increased earlier in the nucleus than in the cytoplasm. In DRG, Gli1 in the nucleus significantly increased from postoperative day 3, and in the spinal cord, it increased from postoperative day 5. Both were maintained at a high level until day 21 (Figure 1(c)). These results demonstrated that hedgehog signaling was significantly activated in the nociceptive pathway after TCI treatment and that Shh signaling was activated earlier in DRG than it was in the spinal cord.

Expression and activation of Shh signaling after TCI treatment. (a) Sample western blot analysis and data summary (n = 4 in each group) showing time-dependent increased expression of Shh, Ptch1, Smo, and Gli1 in dorsal root ganglion (DRG) of rats. (b) Sample western blot analysis and data summary (n = 4 in each group) showing time-dependent increased expression of Shh, Ptch1, Smo, and Gli1 in spinal cord (SC) of rats. (c) Sample western blot analysis and data summary (n = 4 in each group) showing time-dependent increased expression of Gli1 in nuclear extracts from DRG and SC. (d) ELISA assay with concentrations of Shh in ipsilateral tibia lavage fluid of different groups. (e) ELISA assay with concentrations of Shh in ipsilateral sciatic nerve after TCI treatment. (f) ELISA assay with concentrations of Shh in ipsilateral sciatic nerve after CCI treatment. (d) and (e) Samples (n = 10) were collected at postoperative day 7. *P < 0.05, **P < 0.01 vs. Sham group. DRG: dorsal root ganglion; TCI: tumor cell implantation; Shh: sonic hedgehog.
To explore where the Shh protein was released, the concentrations of Shh in tibia lavage fluid and sciatic nerve were tested. The ELISA assay showed that the concentration of Shh significantly increased in the tumor-bearing tibia lavage fluid and sciatic nerve, ipsilateral to TCI (Figure 1(d) and (e)). Further, to clarify whether nerve injury could increase Shh protein release, a CCI model was used. As expected, after CCI treatment, the concentration of Shh increased significantly in the suffering sciatic nerves (Figure 1(f)). These results suggest that both tumor growth and tumor-induced nerve injury could result in Shh protein increase and release.
Inhibition of hedgehog signaling prevented and suppressed generation and persistence of bone cancer pain
According to previous studies,2–4 bone cancer pain is behaviorally characterized by mechanical allodynia, thermal hyperalgesia, spontaneous pain, and movement-evoked pain. To determine the role of hedgehog signaling in bone cancer pain, cyclopamine, a hedgehog signaling inhibitor, was used. In TCI-treated rats, the repeated administration of cyclopamine (10 µg/rat) in the early phase (on postoperative days 2, 3, and 4) significantly delayed the production of mechanical allodynia and thermal hyperalgesia (Figure 2(a) and (b)). The same dose of cyclopamine was administered in the late phase (on postoperative days 7, 8, and 9) and had long-lasting inhibitory effects on the persistence of mechanical allodynia and thermal hyperalgesia (Figure 2(c) and (d)). This analgesia lasted approximately four days after terminating the third injection (Figure 2(c) and (d)). Cyclopamine did not alter normal pain thresholds in sham rats or result in any obvious side effects or animal death. In addition, cyclopamine administration significantly suppressed the established spontaneous pain manifested as guarding and flinching (Figure 2(e) and (f)). In addition, movement-evoked pain manifested as palpation guarding and reduced limb use (Figure 2(g) and (h)). These results demonstrated that inhibiting hedgehog signaling can prevent and suppress the induction and persistence of TCI-induced behavioral hypersensitivity. These findings indicated an important role for hedgehog signaling in the development of bone cancer pain.
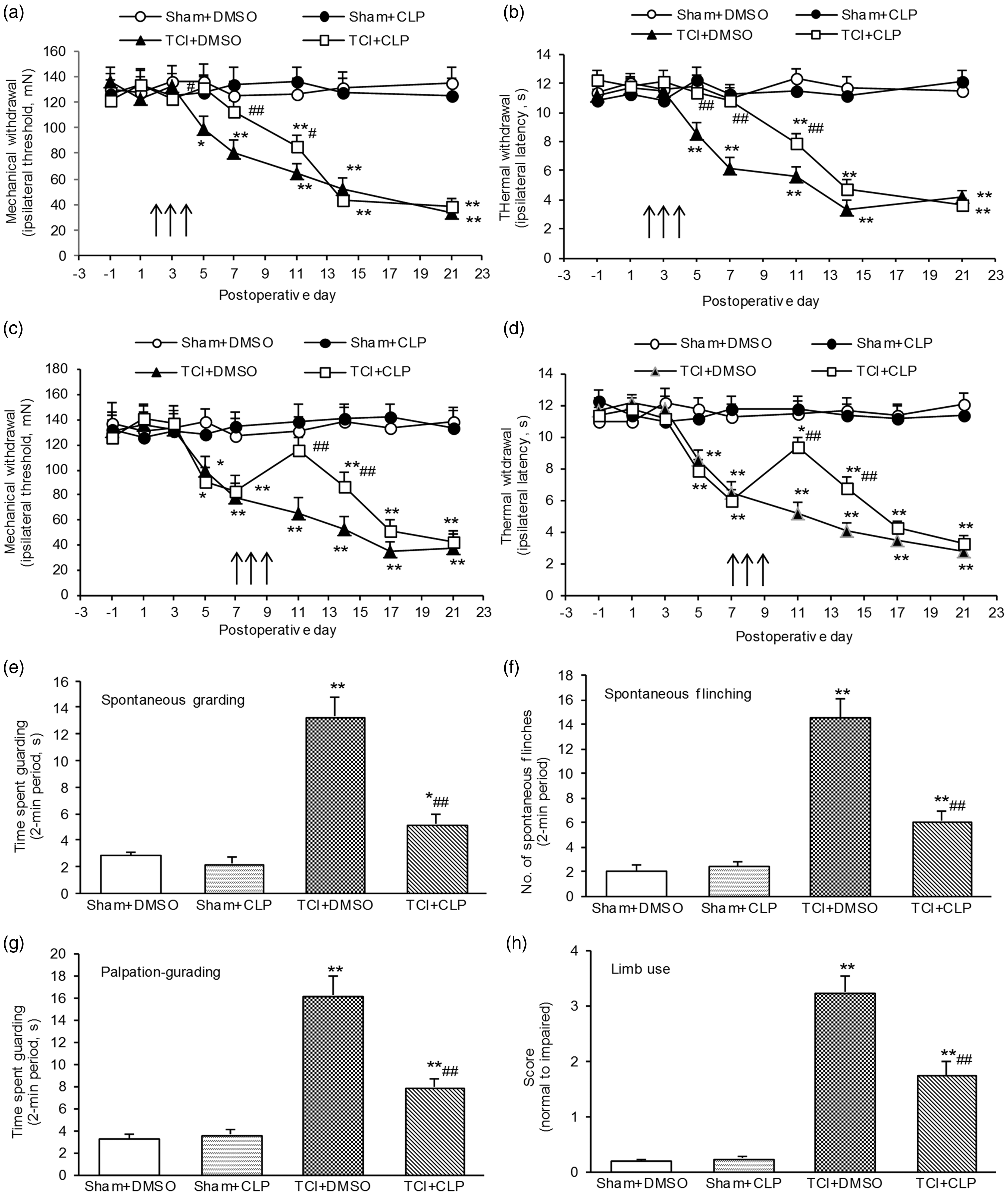
Inhibition of Shh signaling pathway prevented and suppressed bone cancer-induced pain behavior. (a) and (b) Spinal administration of Shh signaling inhibitor cyclopamine (10 µg/rat) in the early stage (postoperative days 2, 3, and 4) obviously prevented TCI-induced mechanical allodynia (a) and thermal hyperalgesia (b). (c) and (d) Spinal administration of Shh signaling inhibitor cyclopamine (10 µg/rat) in the late stage (postoperative days 7, 8, and 9) obviously suppressed and reversed TCI-induced mechanical allodynia (c) and thermal hyperalgesia (d). (e) to (h) Inhibition of Shh signaling pathway suppressed TCI-induced spontaneous pain (e) and (f) and movement-evoked pain (g) and (h). Cyclopamine was administered at postoperative days 7, 8, and 9 (e) to (h). Behaviors were tested at postoperative day 10. Eight rats were included in each group. *P < 0.05, **P < 0.01 indicate significant differences compared with Sham + DMSO group. #P < 0.05, ##P< 0.01 indicate significant differences compared with TCI + DMSO group. TCI: tumor cell implantation.
Inhibition of hedgehog signaling suppressed TCI-induced neurochemical alterations in spinal cord
N-methyl-D-aspartate (NMDA) receptors have a well-developed role in neural plasticity and various pain states. Consistent with previous studies,2,18,19 TCI treatment significantly increased the levels of p-GluN2B, p-CaMKII, and p-CREB. Repetitive administration of cyclopamine (10 µg/rat, i.t. once per day on postoperative days 7, 8, and 9, respectively) significantly inhibited the expression of these molecules in both DRG (Figure 3(a)) and the spinal cord (Figure 3(b)). These results suggested that Shh signaling may contribute to bone cancer pain by regulating synaptic plasticity via NMDA receptor and the subsequent Ca2+-dependent signaling.

Activation of Shh signaling contributes to activation of GluN2B, CaMKII, and CREB in DRG (a) and spinal cord (b) after TCI treatment. In vivo repetitive administration of cyclopamine (10 µg, i.t.) inhibits the phosphorylation of GluN2B, CaMKII, and CREB. Tissues were collected 4 hr after the last injection (n = 4 each group). *P < 0.05, **P < 0.01 indicate significant differences compared with Sham group. #P < 0.05, ##P< 0.01 indicate significant differences compared with TCI group. DRG: dorsal root ganglion; TCI: tumor cell implantation.
Inhibiting hedgehog signaling suppressed TCI-induced intracellular Ca2+ activity
Increases in the intracellular concentration of Ca2+ ([Ca2+]i) reflect neuronal excitation. The activation of hedgehog signaling in DRG and spinal cord after TCI and that inhibiting hedgehog signaling both attenuated TCI-induced hypersensitivity (Figures 1 and 2) and NMDA receptor activation (Figure 3). Thus, we continued to examine the role of hedgehog in TCI-induced, pain-related, increased intracellular Ca2+ activity. The in vitro bath application of a small dose (50 ng/ml) of Shh peptide, Shh-IgG, an exogenous hedgehog signaling activator, did not have a significant effect on [Ca2+]i in small- and medium-sized neurons in the intact DRG from naïve rats (Figure 4(a), left). However, in DRG collected from TCI rats, the in vitro bath application of the same dose (50 ng/ml) of Shh-IgG resulted in a rapid, transient increase in [Ca2+]i, followed by a sustained elevation of [Ca2+]i in small- and medium-sized neurons. (Figure 4(a), middle). Pre-application of cyclopamine (5 µM) significantly reduced the Shh-IgG-induced [Ca2+]i increase (Figure 4(a), right). Figure 4(b) summarizes the data for [Ca2+]i in different groups. Further, to confirm the role of Shh signaling in the activity of intracellular Ca2+, intact DRG from naïve rats were used. The results showed that the transient perfusion of large doses of Shh-IgG (500 ng/ml) effectively resulted in a transient increase in [Ca2+]i in small- and medium-sized neurons (Figure 4(c)). The Shh-IgG-induced increase in [Ca2+]i in naïve DRG was significantly prevented by pretreatment with cyclopamine (5 µM), a Shh signaling inhibitor (Figure 4(d)). BR-A was used to confirm cellular viability. To explore the possible mechanism of Shh signaling in regulating intracellular Ca2+, we investigated the phosphorylation of the NMDA receptor. Activation of the NMDA receptor leads to an influx of Ca2+. The results showed that after Shh-IgG (500 ng/ml) application, the phosphorylated GluN2B increased significantly in the DRG of naïve rat (Figure 4(e)). In addition, the down-stream Ca2+-dependent signal molecules CaMKII and CREB were activated. The phosphorylation of CaMKII and CREB was also significantly increased after Shh-IgG treatment. However, pretreatment with cyclopamine (5 µM) significantly inhibited the Shh-IgG-induced activation of GluN2B and subsequent Ca2+-dependent signaling (Figure 4(e)).
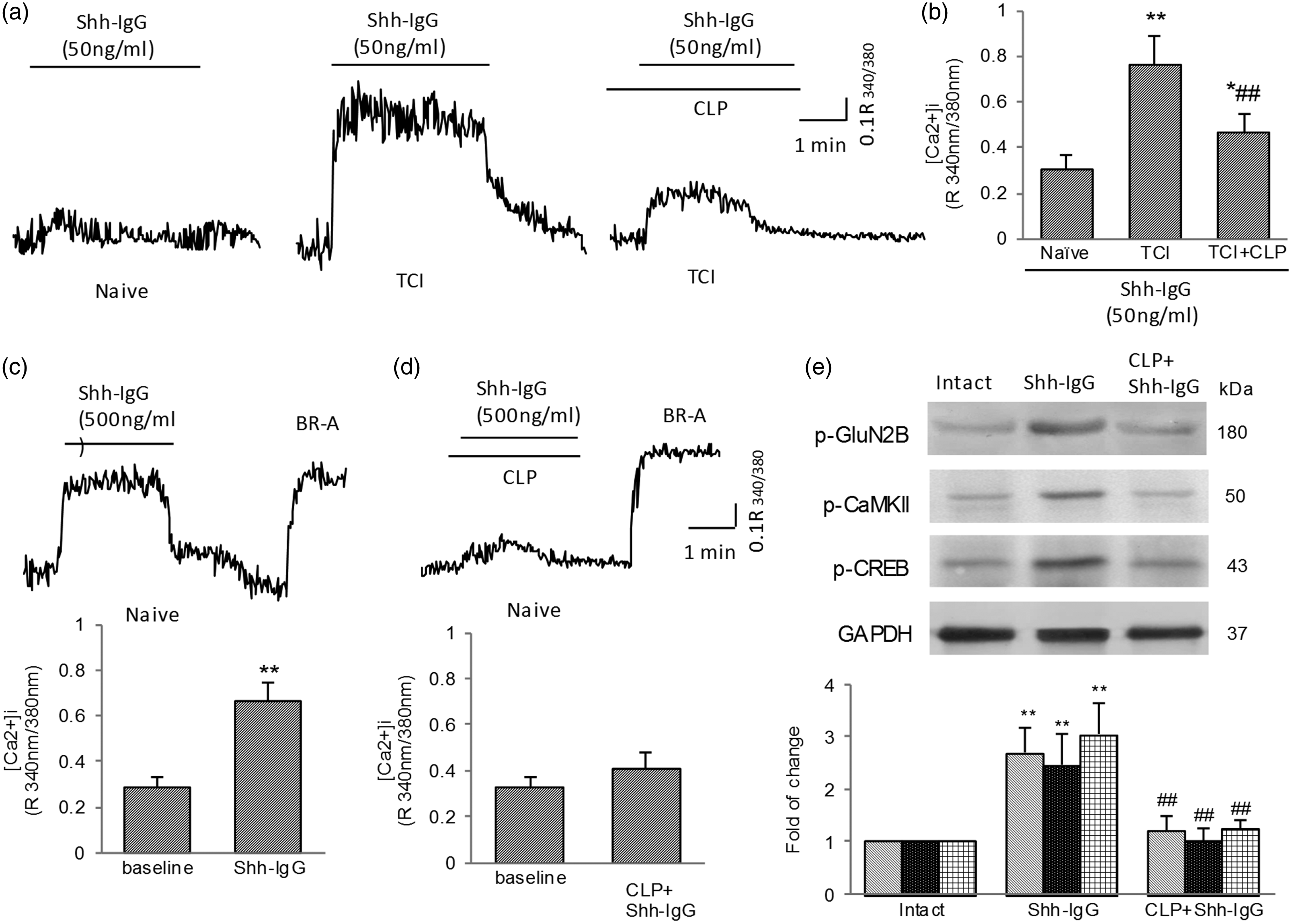
Activation of Shh signaling contributes to increased activity of intracellular Ca2+ in DRG. (a) and (b) In vitro bath of small dose of Shh-IgG (50 ng/ml) failed to increase [Ca2+]i activity in DRG neurons (n = 10) from naïve animals but significantly increased [Ca2+]i activity in TCI-DRG neurons (n= 22). Pretreatment with cyclopamine (5 µM) reduced Shh-IgG (50 ng/ml)-induced increase in [Ca2+]i activity in the DRG neurons (n = 25) from TCI rats. (a) Representative recordings of [Ca2+]i from naïve and TCI-treated, small- and medium-sized DRG neurons. (b) Data summary showing effect of cyclopamine on Shh-IgG-induced activity of [Ca2+]i in TCI-treated DRG neurons. (c) Large dose (500 ng/ml) of Shh-IgG treatment increased [Ca2+]i in intact DRG neurons (n = 30) from naïve rats. (d) Pretreatment with cyclopamine prevented Shh-IgG-induced [Ca2+]i increase in naïve DRG neurons (n= 30). BR-A was used to confirm cellular viability. (e) Western blot analysis showed that Shh-IgG application increased the phosphorylation of GluN2B, CaMKII, and CREB in DRG, which could be inhibited by pretreatment with cyclopamine. Six samples (DRGs) were included in each group. Samples were collected 1 hr after in vitro bath. *P < 0.05, **P < 0.01 indicates significant differences compared with naïve group (b), baseline (c), or intact group (e). ##P < 0.01 indicates significant differences compared with TCI group (b) or Shh-IgG group (e). DRG: dorsal root ganglion; TCI: tumor cell implantation; Shh: sonic hedgehog.
Inhibition of hedgehog signaling reduced TCI-induced hyperexcitability of DRG neurons
Action potential threshold current (APTC) and repetitive discharges were used to reflect neuronal excitability. The lowered APTC and increased repetitive discharges indicate increased DRG neuronal excitability. Examples of neuronal excitability recorded from the prepared DRGs of different groups are shown in Figure 5. Consistent with a previous study, 20 after TCI treatment, the APTC of TCI-treated DRG neurons decreased significantly (∼ 0.25 nA in TCI + DMSO group vs. ∼0.5 nA in Sham group) (Figure 5(a) and (b)), whereas the repetitive discharges greatly increased. The number of spikes increased significantly after TCI treatment (Figure 5(c) and (d)), demonstrating that the excitability of TCI-treated DRG neurons increased markedly. In vitro bath application of cyclopamine (5 µM) significantly and greatly suppressed the increased neural excitability by reversing the decreased APCT and reducing intracellular depolarizing current-induced repetitive discharges (Figure 5(a) to (d)). To confirm the role of Shh signaling in regulating neuronal excitability, intact DRG neurons from naïve animals were used. As expected, the intact DRG neurons from naïve animals were also excited by Shh-IgG administration. The APCT decreased significantly (from ∼0.48 nA to ∼0.21 nA) (Figure 5(e) and (f)), and the repetitive discharges increased markedly (Figure 5(g) and (h)) after Shh-IgG (500 ng/ml) application. Shh-IgG–induced hyperexcitability was prevented by pre-application with the Shh signaling inhibitor cyclopamine. Taken together, these results demonstrate that activation of Shh signaling plays an essential role in the hyperexcitability of DRG neurons during bone cancer pain.
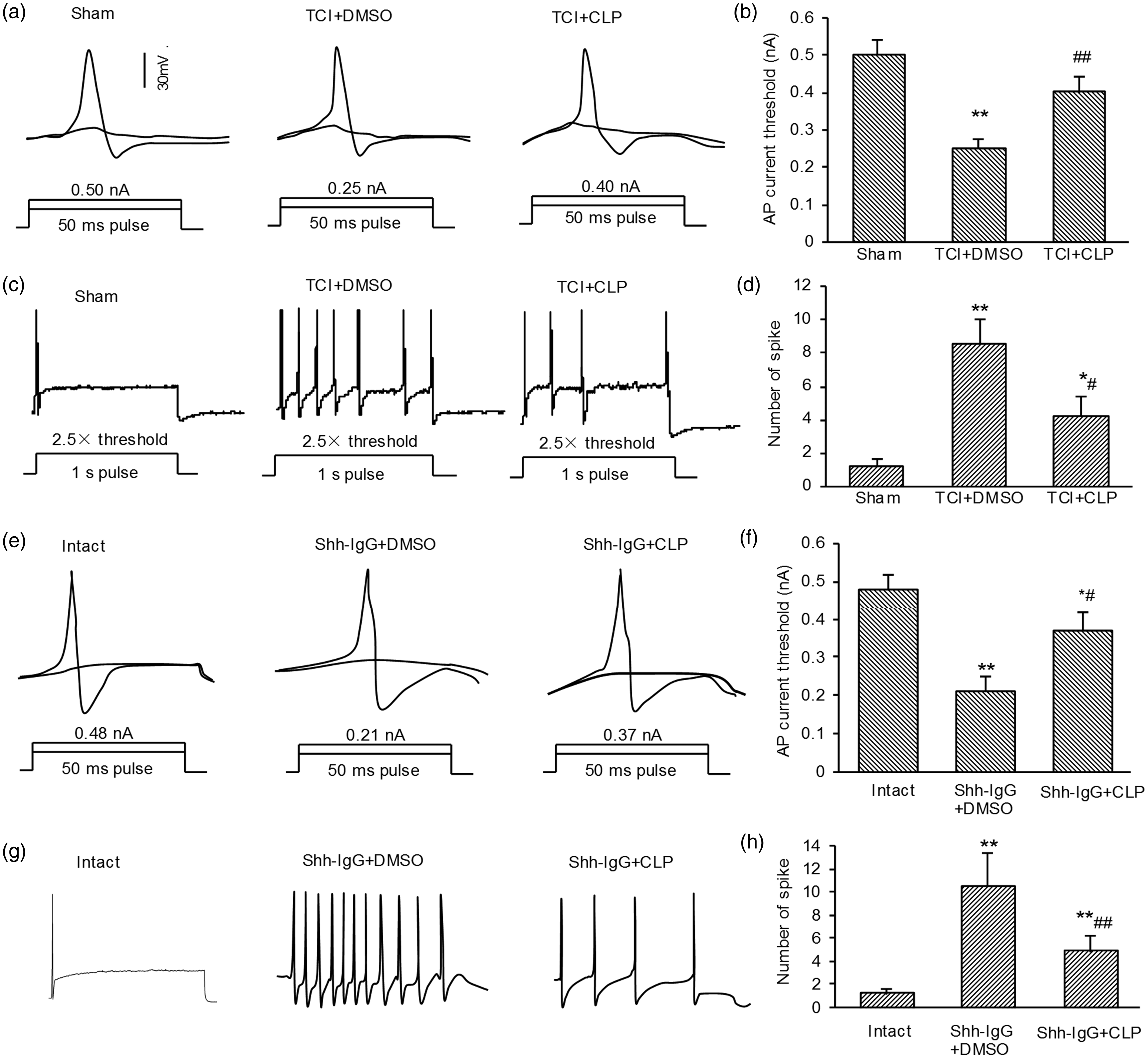
Electrophysiological alteration of excitability of dorsal root ganglion (DRG) neuronal somata after treatment with Shh-IgG, cyclopamine, and TCI, respectively. (a) and (e) Representative neural responses recorded with whole-cell patch-clamp electrodes under current clamp during test sequence, used to determine action potential threshold. Only two of the depolarizing 50-ms pulses (bottom) and corresponding responses (top) are illustrated in each case. (c) and (g) Representative neural discharge patterns evoked by depolarizing current with strength of 2.5 × threshold at 1 s. (b), (d), (f), and (h) Data summary showing effects of Shh signaling inhibition on hyperexcitability of DRG neurons after TCI treatment. (a) to (d) Samples collected at postoperative day 7. (e) to (h) Samples collected 1 hr after in vitro bath. Shh-IgG: 500 ng/ml; CLP: 5 µM (in vitro bath application). Numbers of neurons included in each group: sham = 16, TCI =25, TCI + CLP = 20. Intact = 20, Shh-IgG + DMSO = 25, Shh-IgG + CLP = 25. *P < 0.05, **P < 0.01 indicate significant differences compared with Sham group (b) and (d) or Intact group (f) and (h). #P < 0.05, ##P < 0.01 indicate significant differences compared with TCI + DMSO group (b) and (d) or Shh-IgG + DMSO group (f) and (h). DRG: dorsal root ganglion; TCI: tumor cell implantation; Shh: sonic hedgehog.
Discussion
Our present study reveals the critical role of Shh signaling in the induction and maintenance of bone cancer pain and provides a new understanding of the pathogenesis of bone cancer pain. TCI treatment induced a rapid-onset, long-lasting increase and activation of Shh signaling in both DRG and the spinal cord. Spinal blocking of the Shh signaling pathway effectively prevented and suppressed the induction and persistence of bone cancer pain. The underlying mechanisms of Shh signaling in bone cancer pain regulation may contribute to the regulation of neuron excitability. Blocking Shh signaling reduced the TCI-induced hyperexcitability of the DRG neurons. In addition, exogenous Shh could increase the activity of intracellular Ca2+, and this effect was inhibited by pretreatment with a Shh signaling inhibitor. Taken together, these results demonstrate that Shh signaling plays an important role in bone cancer pain and that Shh signaling contributes to bone cancer pain by regulating the excitability and intracellular Ca2+ activity of DRG neurons. This study suggests that Shh signaling may be a potential target for relieving bone cancer pain.
Shh signaling is known to be important for various developmental processes. Research from past decades has shown a dysregulation of Shh signaling in certain diseases and disorders; e.g., abnormally low levels of Shh signaling are responsible for diverse neurodevelopment disorders, 21 and the constitutive activation of this pathway underlies the occurrence of several cancers. 21 In addition, Shh signaling activation can increase the size of presynaptic terminals at the hippocampal neuron synapses. 9 The ability of the Shh signaling pathway to regulate neuronal connectivity and synaptic plasticity both during and after development has been observed in many parts of the nervous system.22–24 Recently, Shh signaling was reported to be required for modulating nociception and analgesia.11,12,25 In the present study, we provide evidence that supports a new hypothesis that Shh signaling may contribute to the development of bone cancer pain by regulating neuron excitability. TCI caused dramatic, time-dependent changes in the protein expression of Shh signaling in DRG and the spinal cord. Here, we chose L4-L5 DRG and segments of the spinal cord because with tumor growth in the tibia intramedullary space, the nociceptive stimulations were transmitted along the peripheral fibers, especially the sciatic nerve, to the DRG and spinal cord. The sciatic nerve is formed from the L4 to S3 segments of the sacral plexus. The L4-L5 segment of spinal cord is thicker, and the L4-L5 DRG is bigger, whereas the segments of spinal cord blow L6 are too thin, and the DRGs below L6 are too small. To investigate the changes in the corresponding spinal cord induced by bone cancer, we chose the L4-L5 segment. We found that the greatly increased expression of Shh in DRG occurred prior to the onset of behaviorally expressed pain after TCI treatment and that the activation of Shh signaling in spinal cord was well correlated with the timing patterns of TCI-induced bone cancer pain. Spinal administration of the Shh signaling inhibitor significantly prevented and suppressed the production and persistence of bone cancer pain. These results suggest that Shh signaling activation in DRG may be the trigger of bone cancer pain. To explore the sources of increased Shh protein, we further detected the concentrations of the Shh protein in tumor-bearing tibia and the sciatic nerve. The results showed that after TCI treatment, the Shh concentration increased significantly in tumor-bearing tibia and ipsilateral sciatic nerves. To confirm that nerve injury could indeed induce increased Shh release, a CCI model that could induce simple peripheral nerve injury was used. As expected, after nerve injury, the Shh concentration increased significantly in ipsilateral sciatic nerves. Taken together, these results demonstrated that after TCI treatment, with tumor growth, tumor cells and injured nerves produce and release Shh protein and result in an increase in Shh expression in DRG and the spinal cord and then triggers downstream signaling cascades, resulting in intractable pain.
Previous studies suggested potential roles for Shh signaling in synaptic plasticity,5,6 and NMDA receptors have a well-developed role in synaptic plasticity. Thus, we also investigated the changes in the NMDA receptor. Consistent with previous studies,2,19 TCI treatment significantly induced NMDA receptor GluN2B activation in DRG and the spinal cord. Inhibiting Shh signaling activation largely diminished the TCI-induced phosphorylation of GluN2B and the subsequent activation of Ca2+-dependent signaling. As the activation of NMDA receptor led to an influx of Ca2+ and increased intracellular Ca2+ activity, we continued to examine the role of Shh in TCI-induced, pain-related, intracellular Ca2+ activity. Ca2+-dependent electrical activity is the trigger of many intracellular signaling cascades in neurons and plays a key role in various pain states. 26 Shh can elicit Ca2+ spikes in embryonic spinal neurons and thus regulates neuronal differentiation. 27 In the present study, we found that Shh signaling activation obviously increased the activity of intracellular Ca2+ in the intact DRG from TCI rats and that this effect was inhibited by pretreatment of cyclopamine, a Shh signaling inhibitor. Further, to confirm the role of Shh signaling in intracellular [Ca2+]i, DRGs from naïve rats were used. As expected, Shh-IgG application could induce a significant increase in GluN2B activation and intracellular Ca2+ activity, and these effects were inhibited by the pre-administration of cyclopamine. These results demonstrated that Shh signaling may be involved in NMDA receptor activation and in the subsequent Ca2+ influx and Ca2+-dependent signaling activation. After TCI treatment, activated Shh signaling increased GLuN2B activation, which led to Ca2+ influx, enhanced intracellular Ca2+ activity, activated several downstream signal molecules, including CaMKII and CREB,28,29 and ultimately resulted in bone cancer pain.
To confirm the critical role of Shh signaling in bone cancer pain, we also detected the electrophysiological responsiveness of the DRG neurons. Hyperexcitability of nociceptive neurons is likely to be important for pain states.30,31 The activation of sensory neurons with hyperexcitable somata may trigger extra spikes that could contribute to sensitization and hyperalgesia.32,33 Here, we found that the delivery of the Shh signaling inhibitor significantly reduced the TCI-induced hyperexcitability of nociceptive DRG neurons. To confirm the role of Shh signaling in neuronal excitability, the DRG neurons of naïve rats were used. As expected, the pre-administration of CLP significantly inhibited an exogenous Shh-IgG-induced APTC decrease and neuronal discharge increase. These results demonstrated that Shh signaling activation indeed plays an important role in neuronal hyperexcitability. Together, these results provide direct evidence that Shh signaling may contribute to bone cancer pain by regulating nociceptive primary sensory neuron excitability. With the proliferation of cancer cells and tumor growth, Shh signaling was increased and activated in both DRG and the spinal cord, resulting in nociceptive neuronal hyperexcitability, an intracellular Ca2+ activity increase, and Ca2+-dependent signaling activation, leading to painful sequelae. With Shh signaling continuous activation under persistent stimulation, primary and central neurons become sensitized and subsequently produced persistent and intractable bone cancer pain.
In summary, this study reveals a role for Shh signaling in bone cancer pain. The activation of Shh signaling is critical for the induction and maintenance of bone cancer pain. The possible mechanisms of Shh signaling in bone cancer pain regulation may involve the sensitization of DRG neurons and enhanced nociceptive synaptic plasticity by regulating the activity of GluN2B, intracellular Ca2+, and the subsequent Ca2+-dependent signals. Interestingly, the early increase in Shh in DRG suggested that it may be the trigger of bone cancer pain. These findings suggest that targeting the Shh signaling pathway may be an effective approach for treating bone cancer pain.
Footnotes
Author Contributions
SL, YL, and X-XW designed the study. YL performed molecular biological test. X-XW and Z-JS performed the animal behavioral tests. Y-PL performed electrophysiological test. SM performed data collection and statistical analysis. G-LW and G-JL oversaw the execution of the project. SL, YL, and X-XW interpreted the result and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Acknowledgment
The authors thank Zhijiang Huang at University of Texas Southwest Medical Center (UTSW) for his stimulating discussion and help on electrophysiological techniques.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grant from National Natural Science Foundation of China (NSFC-81371242, 81671084), Qing Lan Project of Jiangsu province, Nature Science Foundation of Jiangsu province (BK20161175), “Six One” Project of Jiangsu province (LGY2016039), and Social development project from Lianyungang science and technology bureau (SH1544).