
Abstract
The dorsal horn of the spinal cord is a crucial site for pain transmission and modulation. Dorsal horn neurons of the spinal cord express group I metabotropic glutamate receptors (group I mGluRs) that exert a complex role in nociceptive transmission. In particular, group I mGluRs promote the activation of L-type calcium channels, voltage-gated channels involved in short- and long-term sensitization to pain. In this study, we analyzed the role of group I mGluRs in spinal nociceptive transmission and the possible cooperation between these receptors and L-type calcium channels in the pathophysiology of pain transmission in the dorsal horn of the spinal cord. We demonstrate that the activation of group I mGluRs induces allodynia and L-type calcium channel-dependent increase in nociceptive field potentials following sciatic nerve stimulation. Surprisingly, in a model of persistent inflammation induced by complete Freund’s adjuvant, the activation of group I mGluRs induced an analgesia and a decrease in nociceptive field potentials. Among the group I mGluRs, mGluR1 promotes the activation of L-type calcium channels and increased nociceptive transmission while mGluR5 induces the opposite through the inhibitory network. These results suggest a functional switch exists in pathological conditions that can change the action of group I mGluR agonists into possible analgesic molecules, thereby suggesting new therapeutic perspectives to treat persistent pain in inflammatory settings.
Keywords
Introduction
The role of glutamate as a major excitatory neurotransmitter in spinal nociceptive integration is well established. 1 Glutamatergic activity is mediated by two types of receptors: ionotropic (iGluRs) and metabotropic (mGluRs) receptors. While ionotropic receptors mediate fast excitatory postsynaptic components, mGluRs are coupled to G-protein and mediate long-term changes in nociceptive transmission. The mGluRs are divided into three groups (I, II, and III) based on pharmacology, signal transduction, and sequence homology. 2 In the dorsal horn (DH) of the spinal cord, neuroanatomical and immunohistochemical studies suggest that group I mGluRs are predominantly localized on postsynaptic elements 3 and are involved in the nociceptive transmission.4,5 Indeed, the activation of group I mGluRs increases the excitability of deep dorsal horn neurons (DHNs) in the rat 6 and promotes long-term plasticity. 7 In addition, intrathecal delivery of group I mGluRs agonists induces mechanical and thermal (hot or cold) hypersensitivity as well as spontaneous nocifensive behavior in naive animals.8,9 Similar to group I mGluRs, L-type voltage-gated calcium channels are expressed in the DH and are mainly localized on postsynaptic elements.10,11 LTCs are involved in nociceptive transmission by controlling the short- and long-term plasticity of DHNs.12–15 Finally, it has long been demonstrated that plateau potentials, a mechanism of input/output amplification expressed in deep DHNs, depends on LTCs and is controlled by agonists of mGluRI.6,16–18 In the present study, we analyze the involvement of group I mGluRs and LTCs in nociceptive transmission. We show that in naïve animals, group I mGluRs and LTCs cooperate to amplify nociceptive transmission, whereas in a persistent pain model, group I mGluRs mediate a decrease in nociceptive transmission mediated by the action of subtype mGluR5 on the DH inhibitory network.
Material and methods
Animals
Adult male Sprague Dawley rats (250–350 g) were used in all experiments. Animals were fed and watered ad libitum and kept on 12 h dark/light cycle at room temperature (22℃) with constant humidity. All surgical and experimental protocols were approved by the local ethics committee (ethical approval N°#3765) and conformed to the guidelines of the International Association for the Study of Pain. All efforts were made to minimize the number of animals used and their suffering.
Pain behavior
Mechanical threshold of the nociceptive hind paw withdrawal reflex was measured with a Von Frey electronic device (Bioseb, France). Briefly, rats were individually placed in a cage on a mesh floor. After a 10-min habituation period, constantly increasing pressure was applied to the plantar surface of the hind paw until the animal withdrew its paw. The force applied at the time of withdrawal was recorded, and the response was expressed in grams. Each value was the average of five different stimulations 2-min apart to avoid sensitization of the paw. All behavioral experiments were performed in the morning between 10 a.m. and 12 a.m. All behavioral tests were assessed by the same experimenter blind to the group assignment.
Intrathecal injections
Under isoflurane anesthesia, animals were placed on a cylindric roller to curve the spine. Then, 10 µL (10 mM) of (1S,3R)-ACPD was administered intrathecally with a Hamilton syringe between the L5 and L6 vertebrae. The quality of each injection was ensured by the observation of an injection-induced slight tail-flick reflex. All behavioral tests were blinded. Saline (same volume) was used for control injections.
Field potential recordings
Animals were deeply anesthetized with urethane 20% (1.5 g/kg) administered in a single intraperitoneal injection to induce and maintain anesthesia during the electrophysiology recordings. The experiment was started as soon as there was no longer any reflex. The heat rate was monitored during the experiment. Experiments were stopped when a 10% decrease in heat rate was observed. Colorectal temperature was kept at ∼37℃ with a heating blanket. Two metal clamps were used to set the animal spine in a stereotactic frame (M2E, France) for stability during electrophysiological recordings. Then, a laminectomy was performed at T13-L1 to expose the lumbar part of the spinal cord. The dura mater was removed carefully. A vaseline pool was formed around the exposed spinal segments to ensure that no drug was administered beyond the area of interest. Drugs were applied with a syringe pump (Phymep France). C-fiber-evoked field potentials were recorded in deep lamina of the DH (at a depth range 500 and 1000 µm 12 ) with tungsten microelectrodes (impedance 5 MΩ). Field potentials were recorded with an ISO-DAM-amplifier (low filter: 0.1 Hz to high filter: 0.1 kHz) (World Precision Instruments, USA) in response to electrical stimulation of the ipsilateral sciatic nerve.
The right sciatic nerve was exposed and placed above the two stimulation electrodes. To avoid drying out, the sciatic nerve was covered with paraffin oil. Electrical stimulation was delivered to the sciatic nerve and consisted of single pulses of 0.5 ms duration, at a constant voltage intensity (range: 20–50 V) twice the threshold of onset of C-fiber-evoked field potentials using a master 8 stimulator (AMPI, Israel) connected to an isoflex stimulus isolator (AMPI, Israel). During the experiment, electrical stimulation was applied every 2 min. The C-fiber response can easily be distinguished by its threshold and latency (100–200 ms 19 ). Stable responses of field potentials for half an hour served as control before drug application. Amplitudes of C-fiber-evoked field potentials were measured as the area under the curve in the C-mediated part of the response, i.e., 150 to 300 ms from the stimulation artifact. The average of the first 30 min before drug application was used as the control value, and responses were normalized to it. The mean change during the last 30 min of drug application was used for statistical analysis.
Inflammatory pain model
Inflammation was induced by a subcutaneous injection of 100 µl of complete Freund’s adjuvant (CFA; Sigma Aldrich, Saint Louis, MO) into the dorsal surface of the right ipsilateral hind paw under anesthesia (Vetflurane at 5% for induction and at 2% for maintenance). Control rats received the same volume of 0.9% saline. CFA treatment led to inflammation of the injected paw. A mechanical allodynia was observed from the first day following injection and lasted at least four days. Electrophysiological, biochemical, and behavioral experiments were performed four days after CFA or saline injection.
Drugs
In this study, drugs were applied either directly above the spinal cord during electrophysiology recordings or intrathecally for behavioral tests. Drugs were diluted in saline 0.9% from stock solution and stored at −20℃. Drugs used were group I mGluRs agonist: (1S,3R)-1-amino-cyclopentane-1,3-dicarboxylic acid ((1S,3R)-ACPD, 100 µM, Sigma Aldrich), nifidipine: L-type Ca2+channel blocker (100 µM, Tocris, Bristol, UK), mGluR5 antagonist: 2-methyl-6-(phenylethynyl)-pyridine (MPEP; 100 µM, Tocris), mGluR5 agonist: 2-Chloro-5-hydroxyphenylglycine (CHPG; 100 µM, Tocris), GABA-A receptor antagonist: picrotoxin (10 µM, Sigma Aldrich), and glycinergic receptor antagonist: strychnine (5 µM, Sigma Aldrich).
Tissue preparation
To quantify messenger RNA (mRNA; real-time quantitative polymerase chain reaction (qRT-PCR), see below) and protein levels (Western blot, see below) in the DH, rats were deeply anesthetized with pentobarbital sodium at 150 mg/kg. They were then decapitated, and the lumbar spinal cords were removed. Ipsilateral side of the spinal cord was separated from the contralateral side, and then DH was isolated. Ipsilateral and contralateral DH were then isolated and rapidly frozen in dry ice and stored at −80℃.
RNA extraction and qRT-PCR
RNA extraction from dorsal spinal cord tissue was performed as follows. Frozen ipsilateral or contralateral fragments of the spinal cord were ground in QIAzol lysis Reagent (Qiagen) with a polytron homogenizer (Kinematica). Then, total RNA was purified with the RNeasy Mini kit according to the manufacturer’s instructions (Qiagen). Complementary DNA (cDNA) was synthesized using Maxima First Strand cDNA Synthesis Kit with a mixture of oligo-dT and random hexamer primers (ThermoScientific). PCR amplification was performed on a LightCycler LC480 (Roche) with primer pairs designed to span exon boundaries and to generate amplicons of ∼100 bp. Primer sets for SDHA and mGluR5 were tested by qRT-PCR and gel electrophoresis for the absence of primer-dimer artifacts and multiple products. Triplicate qRT-PCR reactions were done twice for each sample, using transcript-specific primers (600 nM) and cDNA (10 ng) in a final volume of 10 µl. The SYBR Premix Ex-Taq II (Takara) was used according to manufacturer’s instructions. The threshold cycle value of each gene was normalized against that of SDHA. The relative level of expression was calculated using the comparative (2−ΔΔCt) method. 20
Quantitative RT-PCR primers
SDHA Fw: 5′ TGCGGAAGCACGGAAGGAGT 3′ SDHA Rev: 5′ CTTCTGCTGGCCCTCGATGG 3′ mGluR5 Fw: 5′ AGACTTGCAACAGTTCTCTGAC 3′ mGluR5 Rev: 5′ AGGGACATCTGCATATTGTGG 3′
Western blot
Ipsilateral DH samples were manually homogenized in a lysis buffer containing a mix of protease inhibitors. Protein concentration was determined using the method of Bradford. For electrophoresis, protein of 20 µg was loaded onto the gel and electrotransferred to polyvinylidene difluoride membrane. The primary antibody against mGluR5 (anti-mGluR5, 1/1000 (Biotechne)) raised in rabbit, and antibody against tubulin used as control (anti-tubulin, 1/10.000 (Biotechne)) raised in mouse. The secondary antibodies were tagged with peroxidase-conjugated (anti-rabbit (1/2000 (Biotechne)) and anti-mouse (1/2000 (Biotechne)) for mGluR5 and tubulin, respectively. Immunoreactivity was detected using enhanced chemiluminescence Western blotting detection reagents (Super Signal West Femto maximum sensitivity substrate, USA). The mGluR5 protein was detected as bands of relative molecular weight of 130 kDa. The difference in protein expression was examined between control and inflamed animals. The levels of expression of the protein of interest in each sample were analyzed by Image Lab software. The amount of protein was calculated relative to the reference protein band (protein of interest/reference protein (tubulin)), then the values were normalized to the mean of the amount of the control group. The results are expressed as percentage of control ± SEM.
Statistical analysis
Statistical analyses were performed using the GraphPad Prism software (GraphPad Software ©, San Diego, CA). A p value <0.05 was considered significant.
Results
Intrathecal (1S,3S)-ACPD injection produces a mechanical allodynia in control condition
Group I mGluRs agonists are known to promote nociception. We therefore first assessed the consequence of intrathecal (1S,3S)-ACPD injection on pain behavior by measuring the pain withdrawal threshold of the hind paw. Animals received a single intrathecal injection of 10 µl of either (1S,3S)-ACPD (10 mM) or saline. Mechanical paw withdrawal threshold was tested 15 and 30 min after injection. (1S,3S)-ACPD induced a mechanical allodynia 30 min after injection characterized by a significant decrease in withdrawal threshold for both the right hind paw (Figure 1(a), 68.8 ± 5.4 g before injection and 35.5 ± 6 g after ACPD, n = 5, p = 0.03, paired t test) and the left hind paw (Figure 1(a), 68.2 ± 4 g before injection and 42 ± 4 g after ACPD, n = 5, p = 0.009, paired t test). By contrast, saline administration had no effect on paw withdrawal threshold (Figure 1(b), 64.3 ± 3.1 g before injection and 54 ± 6.8 g after saline for the right paw and 65.6 ± 6 g before and 51.5 ± 3.2 g after saline = 5, p = 0.07 and = 0.06, paired t test). To determine whether (1S,3S)-ACPD was responsible for this effect, we performed a two-way analysis of variance to compare the percentage of change in paw withdrawal threshold after either saline or (1S,3S)-ACPD injection (Figure 1(c)). Paw withdrawal threshold of rats injected with (1S,3S)-ACPD was significantly lower than paw withdrawal threshold of saline injected rats for both the right and the left paw (Interaction, F = 7,895, p < 0.01, Bonferroni posttest ACPD vs saline, p < 0.001). Therefore, the decrease in mechanical threshold induced by group I mGluRs agonist suggests a possible increase in nociceptive transmission in the DH.
Effect of (1S,3S)-ACPD on mechanical withdrawal in rats. (a) Intrathecal injection of (1S,3S)-ACPD induces a significant decrease in mechanical withdrawal threshold of both paws. (b) Saline injection has no significant effect on paw withdrawal threshold. (c) Percentage of control of paw withdrawal threshold 15 and 30 min after (1S,3S)-ACPD injection.
Effect of (1S,3S)-ACPD induces an increase in C-fiber-evoked field potentials in control condition
To test whether the decrease in paw withdrawal threshold induced by the activation of group I mGluRs was due to central changes in DH nociceptive transmission, we measured C-fiber-evoked field potentials in the spinal DH of anesthetized adult rats. Potentials were evoked by electrical stimulation of the sciatic nerve with single test pulses. After 30 min of saline superfusion, a solution of (1S,3S)-ACPD was applied at the recording site. (1S,3S)-ACPD induced a significant increase in C-fiber-evoked field potentials 30 min after the drug application (Figure 2(a) and (d); the percentage of change in C-fiber-evoked field potentials was 16.2 ± 5.6%, p < 0.01, n = 17, Wilcoxon signed-rank test). Furthermore, group I mGluRs are known to modulate positively L-type calcium channels (LTCs) that promote mechanisms for short- and long-term sensitization to pain. To assess the possible role of LTCs in the increase in C-fiber-evoked field potentials induced by (1S,3S)-ACPD, we applied nifedipine to block LTCs. When nifedipine was co-applied with (1S,3S)-ACPD, (1S,3S)-ACPD did not change C-fiber-evoked field potentials (Figure 2(b) and (d); the percentage of change in C-fiber-evoked field potentials was −6.33 ± 5.31%, p = 0.29, n = 5, Wilcoxon signed-rank test), while nifedipine alone had no effect on the response of C-fiber-evoked field potentials (Figure 2(c) and (d); the percentage of change in C-fiber-evoked field potentials was 0.95 ± 15.12%, p = 0.95, n = 5, Wilcoxon signed-rank test). Thus, the increase in C-fiber-evoked field potentials by (1S,3S)-ACPD required the activation of LTCs in control conditions.
Facilitatory effect of (1S,3S)-ACPD on the C-fiber-evoked field potentials in dorsal horn of spinal cord. (a) Sixty-minute (1S,3S)-ACPD superfusion induces significant increase in C-fiber-evoked field potentials in control animals (above, raw C-fiber field potentials, scale: 100 ms). (b) Co-superfusion of (1S,3S)-ACPD and nifedipine suppresses facilitatory effect of (1S,3S)-ACPD alone (above, raw C-fiber field potentials, scale: 100 ms). (c) Nifedipine alone did not change C-fiber-evoked field potentials (above, raw C-fiber field potentials, scale: 100 ms). (d) Average of changes induced by (1S,3S)-ACPD (black histogram), (1S,3S)-ACPD and nifedipine (gray bar), and nifedipine (white histogram).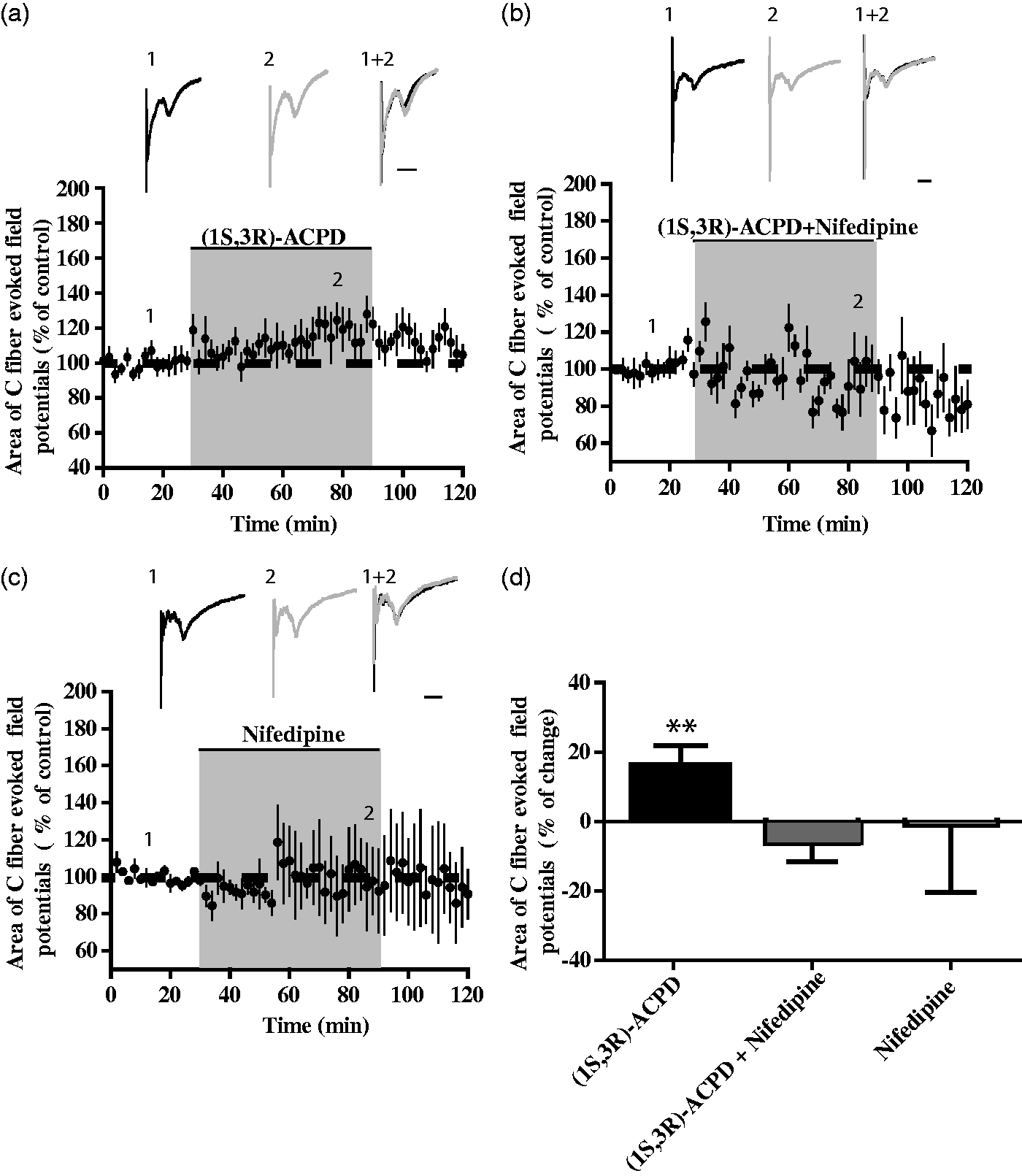
Increase in paw withdrawal threshold induced by (1S,3S)-ACPD in inflamed paw
Intraplantar injection of CFA in the right paw of the animal led to an ipsilateral hypersensitivity characterized at four days postinjection by a marked lower paw withdrawal threshold in the inflamed paw than in the contralateral paw (Figure 3(a), (before injection), 34.5 ± 3.2 g inflamed paw and 64.4 ± 3.3 g contralateral paw, n = 5, p = 0.0015, paired t test). Intrathecal saline injection did not modify this difference (Figure 3(a), (after injection), 37.3 ± 5.1 g inflamed paw and 71 ± 4.5 g contralateral paw, n = 5, p = 0.0046, paired t test). Unexpectedly, intrathecal (1S, 3R)-ACPD suppressed the difference between the ipsilateral and contralateral paw (Figure 3(b), (right part), 50.3 ± 4.8 g inflamed paw and 61.1 ± 3.7 g contralateral paw, p = 0.24, n = 5, paired t test). To confirm this result, we analyzed the percentage of change induced by (1S, 3R)-ACPD in each paw and compared to saline (Figure 3(c)). Thirty minutes after (1S, 3R)-ACPD injection, although not significant as compared to the control situation, paw withdrawal threshold in the inflamed paw was slightly increased, whereas the paw withdrawal threshold in the contralateral paw was slightly decreased. Consequently, there was a statistical difference in the effect of (1S, 3R)-ACPD in the two paws (Figure 3(c), two-way analysis of variance, F = 3,26, p < 0.01, Bonferroni posttest, right paw vs. left paw, p < 0.05).
Unilateral CFA injection induces mechanical allodynia in ipsilateral paw that is partially suppressed by (1S,3S)-ACPD. (a) Four days after CFA injection, paw withdrawal threshold is significantly decreased in ipsilateral paw compared to contralateral paw. (b) After (1S,3S)-ACPD intrathecal injection, the difference between paws is no longer present. (c) Percentage of change in paw withdrawal threshold after either saline or (1S,3S)-ACPD intrathecal injection. Saline did not modify paw withdrawal threshold in either inflamed or noninflamed paw. (1S,3S)-ACPD had a significant opposite effect on both paws, i.e., an increase in paw threshold in inflamed paw and a decrease in contralateral paw.
(1S,3R)-ACPD induces a decrease in C-fiber-evoked field potentials independently from LTCs following inflammation
After CFA injection, the inflamed paw withdrawal threshold was differently modulated by group I mGluR agonists. We next wondered whether nociceptive transmission in the DH was also affected. Indeed, (1S,3R)-ACPD produced a significant decrease in C-fiber-evoked field potentials (Figure 4(a) and (d); the percentage of change in C-fiber-evoked field potentials was −18.21 ± 6.23%, n = 7, p < 0.05, Wilcoxon signed-rank test). These results show that the modulation of nociceptive transmission by group I mGluRs agonists depends on the pathophysiological context, suggesting a plasticity group I mGluRs in the DH after inflammation. We next wanted to know whether the inhibition induced by (1S,3R)-ACPD was dependent on LTCs. To this end, we applied (1S,3R)-ACPD and nifedipine concomitantly. C-fiber-evoked field potentials were decreased in the presence of nifedipine, suggesting that the inhibitory effect of group I mGluRs agonist in an inflammatory setting was independent of LTCs (Figure 4(b) and (d); the percentage of change in C-fiber-evoked field potentials was − 19.13 ± 3.005%, n = 7, p < 0.05, Wilcoxon signed-rank test). Nifedipine alone did not influence DH potentials elicited by C-fiber stimulation (Figure 4(c) and (d); the percentage of change in C-fiber-evoked field potentials = −0.22 ±6.31%, n = 7, p = 1, Wilcoxon signed-rank test).
(1S,3S)-ACPD an inhibition of C-fiber field potentials in CFA-injected paw. (a) (1S,3S)-ACPD superfusion significantly reduced C-fiber field potentials (above, raw C-fiber field potentials, scale: 100 ms). (b) Co-superfusion of (1S,3S)-ACPD and nifedipine did not change inhibition induced by (1S,3S)-ACPD (above, raw C-fiber field potentials, scale: 100 ms). (c) Nifedipine alone had no effect on C-fiber field potentials (above, raw C-fiber field potentials, scale: 100 ms). (d) Average of percentage of change following (1S,3S)-ACPD (black histogram), (1S,3S)-ACPD and nifedipine (gray histogram) and nifedipine (white histogram).
Inflammation induced a switch from mGluR1 to mGluR5 sensitivity to (1S,3R)-ACPD
Group I mGluRs include mGluR1 and mGluR5. We hypothesized that peripheral inflammation alters mGluR5 expression. Indeed, mGluR5 is a well-known peripheral receptor involved in changes induced by different pain models. To check this hypothesis, we used qRT-PCR to measure the change in mGluR5 mRNA in the DH. We compared the levels of mRNA expression in both saline and CFA animals and compared the ipsilateral and contralateral parts of the DH. Inflammation did not alter mGluR5mRNA expression (Figure 5(a), p > 0.05, n = 6, Test de Fisher + Student’s test, Figure 5(a)). Therefore, we assessed the possibility of posttranslational modification at the protein level. To this end, we used Western blot to compare the level of mGluR5 proteins in the ipsilateral DH of saline and CFA animals. We observed a significant increase in mGluR5 protein levels in the ipsilateral part of the DH in CFA animals as compared to saline ones (Figure 5(b), 41.7 ± 10.25% of increase in protein expression in inflammatory condition, p = 0.0096, Wilcoxon signed-rank test, n = 6). We then assessed whether a change in mGluR5 protein level could account for the inhibition of C-fiber field potentials induced by (1S,3R)-ACPD in inflammation.
Posttranslational increase in mGluR5 expression after CFA-induced inflammation. (a) qRT-PCR of mGluR5 mRNA in saline versus CFA dorsal horn of spinal cord. mRNA levels not modified. (b) Western blot of mGluR5 proteins demonstrates significant increase in mGluR5 protein levels as compared to saline injection (B1). CFA: complete Freund’s adjuvant.
mGluR5 mediates a decrease in C-fiber-evoked field potentials following inflammation
To test whether this upregulation influences nociceptive transmission, we applied CHPG, a specific agonist for mGluR5 receptors. CHPG produced a significant decrease in C-fiber-evoked field potentials in the inflammatory condition (Figure 6(a) and (d); the percentage of change in C-fiber-evoked field potentials was −28.49 ±6.18%, n = 9, p < 0.01, Wilcoxon signed-rank test). By contrast, MPEP, an antagonist of mGluR5, had no effect on C-fiber-evoked field potentials (Figure 6(d), −11 ± 6%, n = 8, p = 0.14, Wilcoxon signed-rank test). Finally, MPEP prevented the decrease in C-fiber-evoked field potentials in the presence of (1S,3R)-ACPD (Figure 6(b) and (c); the percentage of change in C-fiber-evoked field potentials was −4.77 ± 3.59%, n = 6, p = 0.25, Wilcoxon signed-rank test). These results demonstrate that the decrease in C-fiber-evoked field potentials induced by (1S,3R)-ACPD is mediated by the activation of mGluR5.
Inhibition of C-fiber field potentials depends on mGluR5. (a) CHPG, an agonist of mGluR5, induces significant reduction in C-fiber field potentials (above, raw C-fiber field potentials, scale: 100 ms). (b) MPEP, an antagonist of mGluR5, suppresses inhibitory action of (1S,3S)-ACPD (above, raw C-fiber field potentials, scale: 100 ms). (c) Blockade of inhibitory influences by superfusion of picrotoxin and strychnine suppresses inhibitory effect of (1S,3S)-ACPD (above, raw C-fiber field potentials, scale: 100 ms). (d) Average of percentage of change induced by CHPG (black histogram), MPEP (gray gradient), (1S,3S)-ACPD and MPEP (gray histogram), and (1S,3S)-ACPD and picrotoxin and strychnine (white histogram).
(1S,3R)-ACPD-induced antinociceptive effect in ipsilateral DH is mediated by local inhibitory network
Within the spinal cord, the GABA-A and glycine receptors are the two main inhibitory receptors and their activation is crucial for controlling nociceptive transmission. We therefore sought whether the mGluR5 present in inhibitory interneurons could mediate the inhibitory effect of (1S,3R)-ACPD in inflammation. To this end, we blocked inhibition in inflamed rats by applying strychnine and picrotoxin, which inhibit the glycine and GABA-A receptors, respectively, during the application of (1S,3R)-ACPD. Blockade of the inhibitory transmission prevented the decrease in C-fiber-evoked field potentials induced by (1S,3R)-ACPD (Figure 6(c); the percentage of change in C-fiber-evoked field potentials was 2.05 ± 7.04%, n = 7, p = 0.77, Wilcoxon signed-rank test). However, (1S,3R)-ACPD did not induce an increased C-fiber-evoked field potential suggesting a decrease of mGluR1 influence following inflammation. This demonstrates that the increase in mGluR5 in inflamed rats mediated inhibition of C-fiber-evoked field potentials via inhibitory interneurons in the DH.
mGluR5-dependent inhibition is insufficient to counterbalance the mGluR1-mediated potentiation of C-fiber-evoked field potentials in control rats
Finally, we sought whether C-fiber-evoked field potentials were inhibited in control rats. As in inflamed rats, CHPG produced a decrease in C-fiber-evoked field potentials (Figure 7(a) and (c); the percentage of change in C-fiber-evoked field potentials was −21.31 ± 5.77%, n = 11, p < 0.01, Wilcoxon signed-rank test). Finally, to confirm that mGluR5 is not involved in the potentiation of C-fiber-evoked field potentials, we blocked mGluR5 with MPEP and applied (1S,3R)-ACPD. (1S,3R)-ACPD produced an increase in C-fiber-evoked field potentials in the presence of MPEP (Figure 7(b) and (c); the percentage of change in C-fiber-evoked field potential was 33 ± 14.2%, n = 10, p = 0.01, Wilcoxon signed-rank test). These results showed that in control conditions, mGluR5 mediated inhibition of C-fiber-evoked field potentials, but this inhibition was too weak to reverse the potentiation induced by the activation of mGluR1 with (1S,3R)-ACPD. This strongly suggests that the increase in mGluR5 protein levels in inflamed conditions masked the potentiation of C-fiber-evoked field potentials induced by (1S,3R)-ACPD via mGluR1.
Inhibitory effect of mGluR5 is reduced in control conditions. (a) CHPG superfusion induced a reduction in C-fiber field potentials in control conditions (above, raw C-fiber field potentials, scale: 100 ms). (b) Co-superfusion of (1S,3S)-ACPD and MPEP did not modify facilitatory effect of (1S,3S)-ACPD, suggesting that the latter depends on mGluR1 (above, raw C-fiber field potentials, scale: 100 ms). (c) Average percentage of change induced by CHPG (black histogram), ACPD and MPEP (gray histogram).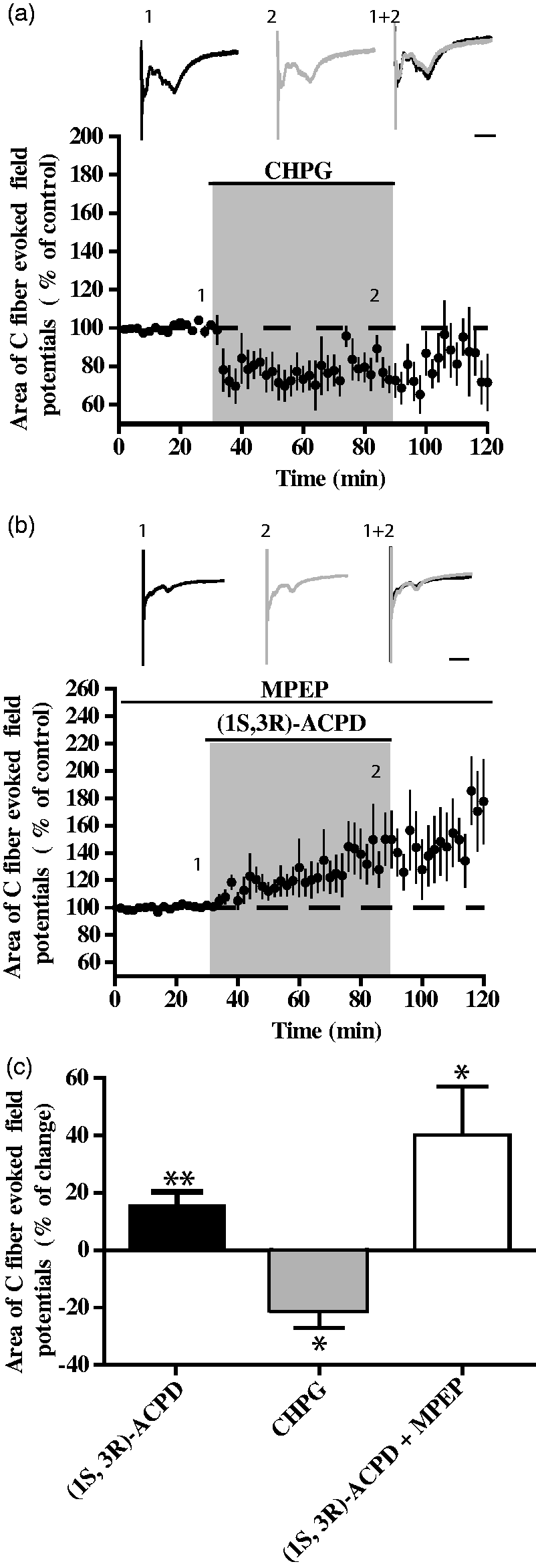
Discussion
The present study investigated the consequence on nociceptive transmission of activating group I mGluRs. We particularly examined the putative cooperation between group I mGluRs and LTCs in the pathophysiology of pain transmission in the DH. We show that inflammation deeply modifies the relative contribution of group I mGluRs to agonist application. The activation of mGluR1 exerts a pronociceptive role in the DH through the activation of LTCs, while mGluR5 exerted an antinociceptive action through the local inhibitory network in the conditions of inflammation (Figure 8).
General mode of action of (1S,3S)-ACPD on C-fiber field potentials in spinal cord in control and CFA-induced inflammation. (1S,3S)-ACPD mainly acts via activation of mGluR1 that mediates activation of L-type calcium channels, while (1S,3S)-ACPD acts via mGluR5 on the inhibitory network in CFA-induced inflammation.
Activation of Group I mGluRs increases nociception via LTCs
By using pain behavior measurements and in vivo electrophysiology, we confirm that the spinal administration of (1S,3R)-ACPD induces an increase in nociceptive transmission. 21 Indeed, intrathecal injection of (1S,3R)-ACPD induced a bilateral decrease in the mechanical paw withdrawal threshold, i.e., a mechanical allodynia. Moreover, spinal application of (1S,3R)-ACPD led to a significant increase in C-fiber-evoked field potentials, thus confirming the spinal action of (1S,3R)-ACPD. These results are in accordance with previous results showing that (1S,3R)-ACPD increases acute hyperalgesia induced by ionotropic glutamate modulators. 22 They are also in line with studies showing that group I mGluRs agonists elicit an increase in the excitability of DHNs.23,24 Group I mGluR is also involved in the long-term potentiation of C-fiber nociceptive field potentials, a spinal mechanism of hypersensitivity.25,26 (1S,3R)-ACPD applied to the DH increases the amplitude of the windup of spinal neurons, a form of short-term sensitization to pain that is considered to be an indicator of central sensitization to pain. 24 By contrast, this windup is blocked by group I mGluRs antagonists. Moreover, (1S,3R)-ACPD increases the amplitude of plateau potentials that sustain the expression of windup.6,18
Both long term potentiation (LTP) and windup depend on the expression of LTCs, which are crucial for neuronal excitability within the DH.12,14,15,27 Here, we show that LTCs mediate the pronociceptive effect of (1S,3R)-ACPD. We also confirm that LTCs alone do not modify acute pain transmission.12,28 This increase in the level of activation of LTCs by group I mGluRs agonist has been well demonstrated in both the dorsal and ventral horn of the spinal cord.18,29 Moreover, this effect is mainly mediated by the subtype mGluR1. 6
In our conditions, the pronociceptive effect of group I mGluRs agonist acting through LTCs was not suppressed by the mGluR5-specific antagonist, suggesting that (1S,3R)-ACPD acts via the subtype mGluR1. In conclusion, our results strongly suggest that (1S,3R)-ACPD elicits an increased nociception via the activation of LTCs in DHNs. However, mGluR1 and LTCs are also expressed in afferent nociceptive C-fibers, and we cannot exclude that this peripheral pathway also participate in controlling nociceptive transmission in the DH (Figure 8).
One of the possible mechanisms of this direct facilitating effect of mGluR1 on LTCs is the phospholipase C (PLC)-IP3 (inositol triphosphate) cascade, which leads to the release of calcium from the intracellular stores that binds to calmodulin, leading to the activation of LTCs, as previously demonstrated.29–31 Group I mGluRs may also have an indirect positive effect on LTCs by inhibiting inward rectifier potassium channels that induce membrane depolarization necessary for the activation of LTCs.18,32
mGluR5-dependent decreases in nociception induced by group I mGluRs agonists in persistent inflammation
Activation of group I mGluRs by (1S,3R)-ACPD reduces spinal nociceptive transmission following CFA-induced inflammation, suggesting a functional change in the expression and/or distribution of the receptors activated by (1S,3R)-ACPD under these pathological conditions. Indeed, intrathecal injection of (1S,3R)-ACPD had an antinociceptive action and decreased the nociceptive field potentials induced by electrical stimulation, specific to the inflamed paw. Such an opposite effect of group I mGluRs agonists under pathophysiological states has already been observed in the excitability of DHNs following intrathecal injection of carrageenan. 33
We show here that this antinociceptive effect of (1S,3R)-ACPD depends on mGluR5 activation. Indeed, after inflammation, we observed a significant increase in mGluR5 protein expression in the ipsilateral DH, a decrease in nociceptive field potential amplitudes following application of mGluR5 agonist, and a lack of effect of (1S,3R)-ACPD when a mGluR5 antagonist was applied. These results are in agreement with previous studies that showed a dose-dependent inhibition of the spinothalamic tracts with a specific agonist of mGluR5. 5 This is surprising given that mGluR5 has been described as being pronociceptive. Indeed, blockade of mGluR5 with the specific antagonist MPEP decreased hypersensitivity induced by partial nerve ligation or carrageenan injection. 34 This pronociceptive role is due to the activation of peripheral mGluR5 rather than spinal ones.34–36 Therefore, we cannot rule out the spatial and functional segregation of mGluR5. Finally, a recent study emphasized the role of intracellular mGluR5 in the DH in a mononeuropathy model. While it is unlikely that these receptors were activated in our conditions, 37 we cannot rule out the possibility that our results are specific to the persistent inflammatory model we used. Finally, our findings demonstrate that under persistent inflammation, group I mGluRs agonists have a prominent spinal antinociceptive effect via activation of mGluR5.
mGluR5-dependent decreases in nociception mediated by spinal inhibitory network
The activation of group I mGluRs has been shown to facilitate both inhibitory and excitatory neurotransmission in the DH. 38 However, nothing is known about a putative cellular segregation of the two mGluR subtypes in this site. We show here that the pronociceptive role of mGluR agonists depends on mGluR1 while antinociception is mediated by mGluR5. Moreover, in the context of persistent inflammation, mGluR5 protein expression is increased, and group I mGluR agonists produce antinociception. Finally, we also show that this effect is blocked by blockade of the local inhibitory network. Therefore, it is likely that mGluR5 is localized on inhibitory interneurons that produce this inhibition of nociceptive transmission (Figure 8). We cannot exclude a decrease of mGluR1 influence in the context of inflammation since group I mGluR agonists did not induce an increase in C-fiber-induced field potentials even under inhibitory network blockade. By contrast, in control conditions, levels of mGluR5 might not be sufficient to overcome the main pronociceptive action of mGluR1. This antinociceptive effect of mGluR5 under persistent inflammation is independent of LTCs, suggesting that mGluR5 acts via other intracellular machinery (Figure 8).
Concluding remarks
In summary, we show in this study that the same group I mGluRs agonist exerts an opposite action on spinal nociceptive transmission depending on the pathophysiological state. In control conditions, a pro-nociceptive action mediated by mGluR1 was obtained via activation of LTCs, while in a setting of persistent inflammation; an mGluR5-dependent anti-nociceptive action was evidenced. The findings also suggest that the mGluR subtypes have a different spatial distribution, mGluR5 being localized on the inhibitory interneurons while mGluR1 is potentially present on both the C-fibers and the relay neurons expressing LTCs. In a context where mGluR5 blockers could potentially be used to treat neuropathic pain patients, our findings have important therapeutic implications since understanding of the origin of pain syndromes is crucial for the appropriate pharmacological management of pain.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the CNRS and the University of Bordeaux, Erasmus Mundus Green-it and Labex “BRAIN”.