
Abstract
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease with manifestations ranging from mild skin symptoms to severe organ and neurological involvement. Thrombosis is a major complication and leading cause of mortality, though its mechanisms remain unclear. Emerging evidence highlights platelets, beyond their hemostatic role, as central players in SLE pathogenesis. This narrative review summarizes platelet-driven pathways in SLE, emphasizing their roles in thrombosis and immune regulation. Platelets interact with immune complexes, complement, and infectious agents, triggering activation and the release of mediators and microparticles. These processes increase circulating autoantigens and promote both thrombosis and autoimmune responses. Understanding these non-hemostatic platelet functions offers new insight into SLE mechanisms and may guide future platelet-targeted therapies.
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease of unclear etiology that primarily affects young women. It exhibits a hypercoagulable condition with a high mortality rate. In this disorder, the immune system mistakenly targets various organs and tissues, producing varied antibodies against nuclear and cytoplasmic antigens that result in organ damage. 1 The accumulation of autoantibodies and autoantigens, mainly DNA and nuclear components in antibody-antigen complexes within the bloodstream and their deposition in various organs and tissues plays a primary role in the pathogenesis of SLE. 2 This process leads to complications like nephritis, arthritis, nervous disorders, and skin rashes. 3 Cardiovascular complications are a primary cause of mortality in SLE, with progressive atherosclerosis and coronary heart disease (CHD) being particularly significant. 4 The global prevalence of SLE ranges from 3 to 517 cases per 100,000 individuals, with a notable gender disparity of 9:1 in favor of women. 5 It is essential to recognize the diverse clinical manifestations of SLE. 6 SLE affects all three blood cell types, leading to anemia of chronic disease (ACD), leukopenia, and mild thrombocytopenia in patients. 7 Mounting pieces of evidence underscore the significant role of platelets and their activation in the pathogenesis of SLE.8,9
This narrative review focuses specifically on platelet activation, platelet–immune interactions, and platelet-derived microparticles in the pathogenesis of SLE, with the aim of synthesizing current mechanistic evidence and clarifying the contribution of platelets to inflammation, thrombosis, and immune dysregulation. While previous reviews have separately explored thrombosis or immune dysregulation in SLE, our work synthesizes these interconnected pathways and highlights how platelets serve as a mechanistic bridge between inflammation and vascular injury. By consolidating emerging findings from proteomics, immunology, and vascular biology, this review identifies current knowledge gaps and proposes directions for future platelet-targeted diagnostic and therapeutic strategies. To gather relevant literature, we performed a targeted search of PubMed, Scopus, Web of Science, and Google Scholar using combinations of the terms ‘systemic lupus erythematosus’, ‘platelets’, ‘platelet activation’, ‘microparticles’, and ‘thrombosis’. Additional articles were identified through cross-referencing key publications. Only English-language studies published up to November 2025 were included.
2. Role of platelets in SLE
Platelets contain three granule types: alpha, dense (delta), and lysosomal. Alpha granules (40–80/platelet) carry P-selectin, von Willebrand factor (vWF), clotting factors, platelet factor 4 (PF4), and growth factors such as platelet-derived growth factor (PDGF) and transforming growth factor beta (TGF-β) for hemostasis and immunity. Dense granules (4–8/platelet) store ADP, ATP, Ca2+, Mg2+, serotonin, and histamine to boost activation. Lysosomes, proteasomes, Golgi remnants, mitochondria, and membrane phospholipids (source of arachidonic acid for thromboxane A2 (TXA2)) further support platelet function. 10
Beyond their role in homeostasis, platelets interact with immune cells, releasing microparticles platelet-derived microparticles (PMP) and inflammatory molecules within their granules, such as PDGF. This interaction extends to innate and acquired immune cells like lymphocytes, macrophages, and dendritic cells, labeling platelets as activators of the acquired immune system. Consequently, these cells are implicated in immune homeostasis.11,12
Given the multifaceted roles of platelets, they affect the pathogenesis of SLE in various ways by participating in inflammation, endothelial dysfunction, thrombosis, immune response, and tissue damage. Activated platelets produce inflammatory mediators, fostering systemic inflammation in SLE through interaction with immune cells, causing vascular dysfunction and tissue damage. In the context of SLE, platelets contribute to an increased thrombotic risk by enhanced accumulation and activation of the coagulation cascade, promoting abnormal platelet activation, adhesion, and aggregation.13,14 Strong evidence supports the assertion that the aberrant response of the innate and acquired immune systems along with cell dysfunction contributes to progressive atherosclerosis in patients with SLE. Factors such as incomplete apoptosis, production of antibodies (anti-Smith, anti-dsDNA, antiphospholipid antibodies (APL), antinuclear antibodies), aberrant neutrophilic response, and type I interferon pathway all contribute to endothelial cell dysfunction. 15
In normal conditions, nuclear antigens are swiftly cleared from the blood after apoptosis. However, in patients with SLE, increased load of apoptosis products and their stability can lead to an inflammatory response. 16 Consequently, irregularities in lymphocyte function, accumulation of antibodies, incomplete clearance of immune complexes, and apoptosis products are characteristics of SLE. 10 The primary cause of organ damage in SLE is the immune system itself as immune complexes persist in blood vessels, causing damage to various organs. 17
Platelets and PMPs play a vital role in the development of fibrosis in organs such as the heart, kidney, lung, and skin. Platelets are a key source of circulating TGF-β that is released upon activation and promotion of fibrosis in the skin and lungs through the induction of fibroblast proliferation, extracellular matrix degradation, and upregulation of profibrotic genes. 18
3. Platelet activation in SLE
Platelet-activating factors in SLE.

In addition to FcγRIIA, immune complexes may activate platelets through TLR4 and TLR7. 9 Platelets express multiple pattern-recognition receptors (TLR2, TLR4, TLR7, TLR9) that detect damage-associated molecular pattern (DAMPs) and PAMPs. Elevated Lipopolysaccharide (LPS) levels activate TLR4 in SLE, as evidenced by CD14 shedding. 29 Upregulated TLR9 on activated platelets correlates with disease activity. 20 Platelet TLR4 engagement promotes neutrophil activation via GPIb–CD18 interactions, driving Neutrophil extracellular traps (NETosis) and release of pro-inflammatory and pro-thrombotic mediators. 9 Studies disagree on whether FcγRIIA signaling (immune-complex–driven) or TLR4/TLR7 pathways (endotoxin/viral RNA–driven) dominate platelet activation in SLE.9,20 This variability reflects differences in patient serology, immune complex composition, and exposure to microbial products.
Platelets also express NOD-like receptors P3 (NLRP3), which responds to mitochondrial reactive oxygen species (ROS), oxidized DNA, and viral RNA by enhancing IL-1β production. 24 Expression of FcαRI (CD89) and FcεR (CD23) offers additional potential routes for activation through IgA- and IgE-containing immune complexes.27,28
Activated platelets contribute to organ damage, nephritis, thrombosis, and cardiovascular disease by promoting interferon production, NETosis, and activation of dendritic cells and lymphocytes. 10 Phosphatidylserine exposure facilitates thrombin generation, enhancing coagulation. 30
Activation of αIIbβ3 integrin further supports aggregation. 31 Type I interferon (IFN) heightens platelet activation through effects on megakaryocyte gene expression, correlating with thrombotic risk. 32
Platelet activation is assessed through phosphatidylserine exposure, CD62P expression, and platelet–monocyte aggregates. 33 These markers are elevated in SLE and contribute to inflammatory amplification and endothelial dysfunction.3,34
The activation pathways described above have clear clinical relevance, as they closely mirror major outcomes in SLE—including thrombosis, nephritis, endothelial injury, and overall disease activity. Markers of platelet activation such as CD62P expression, phosphatidylserine exposure, platelet–leukocyte aggregates, and PC4d deposition correlate with higher SLEDAI scores, vascular events, and renal involvement.31,32,35 Type I IFN–driven platelet priming and persistent immune complex–mediated activation further link platelet biology with vascular risk and flare severity.20,30 Collectively, These mechanistic insights also support the growing interest in platelet-targeted therapy in SLE. Antiplatelet agents (e.g., aspirin, P2Y12 inhibitors), blockers of platelet–leukocyte interactions (e.g., P-selectin inhibition), complement-targeted therapy, and Janus kinase (JAK) inhibition to reduce IFN-driven platelet activation may help modulate both inflammation and thrombotic risk.30,32,36 Targeting PMP-associated pathways—such as tissue factor and phosphatidylserine—represents another potential strategy to limit thrombin generation and endothelial injury.37–39 Overall, platelet activation appears to bridge immune dysregulation with vascular pathology in SLE, underscoring its potential as both a biomarker and a therapeutic target.
4. Platelets and thrombosis in SLE
The risk of thrombosis is significantly increased in patients with SLE, with most recent population-based studies estimating a 4–6-fold higher risk compared to the general population. Earlier cohort studies reported even higher estimates, up to 25–50-fold. Venous thrombosis occurs more frequently than arterial events in patients with SLE.40–42 Notably, young women with SLE have a 50-fold higher risk of myocardial infarction compared to age and sex-matched controls. 8 Common thrombotic events in SLE encompass deep vein thrombosis, arterial thrombosis, pulmonary embolism, coronary artery occlusion, and cerebrovascular events.20,43 However, the relatively infrequent occurrence of thrombotic events over the extended course of the disease poses challenges in conducting prospective studies related to thrombosis in patients with SLE. 44
Studies show that the presence of APL strongly increases the risk of thrombosis in patients with SLE, but about 20% of lupus patients with thrombosis are negative for APL, so there are reasons other than APL for thrombosis in these patients.37,45 Platelets, endothelial cells, and circulating coagulation factors are the primary components of thrombosis. There have been limited studies on the role of aberrant platelet activation in causing thrombotic events in patients with SLE. Chronic platelet activation, which is characterized by phosphatidylserine exposure, secretion of proinflammatory and prothrombotic molecules (e.g., platelet microparticles, cytokines, and chemokines), and interaction with endothelial cells and immune cells, may underlie thrombotic events in patients with SLE, the full mechanism of which remains incompletely understood. 45 Activated platelets attract circulating platelets through the release of mediators like TXA2, ADP, ultra-large multimers of vWF (ulvWF), serotonin, and the production of thrombin, leading to coagulation. 43
The following subsections discuss the major platelet-related factors in the pathogenesis of thrombosis in SLE, each discussed separately to emphasize their unique role in the pathogenic process.
4.1. FcγRIIA
FcγRIIA remains an important mediator of platelet activation in SLE, and increased anti-dsDNA titers—alone or in complexes—promote activation via αIIbβ3, αvβ3, and FcγRIIA. 20 On the other hand impaired immune‐complex clearance by platelets may further contribute to vascular injury. 10 The H131R FcγRIIA polymorphism alters IgG2 affinity and is associated with elevated soluble E-selectin and increased platelet–endothelial interactions. 11 FcγRIIA overexpression in mouse models exacerbates nephritis and induces pulmonary and renal thrombosis. 38
4.2. PMPs
PMPs, namely extracellular vesicles approximately 100-1000 nm in diameter released from the membrane of activated platelets upon stimulation (e.g., collagen or thrombin, ICs, C5-9 membrane attack complex, apoptosis, inflammatory factors such as LPS or viruses), contribute to SLE thrombosis through various mechanisms. PMPs, rich in enzymes, cytokines (e.g., IL-1β, IL-6, IL-8, tumor necrosis factor), microRNA, mRNA, mitochondria, CD41, P-selectin, and high mobility group box 1 (HMGB1), mediate processes such as inflammation, coagulation, vascular endothelial dysfunction, and autoantibody production.39,46 Also the surface of PMPs, abundant with phosphatidylserine, activates the internal coagulation pathway with TF and VII, as well as activating the external coagulation pathway by promoting increased thrombin production. 47 Studies suggest elevated concentrations of PMPs in patients with SLE correlate with disease activity, suggesting their potential as prognostic indicators for diagnosis, treatment, and treatment response monitoring.46,48 Accordingly blocking autoantigens carried by PMPs emerges as a potential strategy for SLE treatment. 49 In spite of this, different studies report varying degrees of association between PMP levels and thrombotic risk.39,47 The inconsistency may be influenced by methodological techniques such as flow cytometry sensitivity, sample handling.
4.3. Serotonin
Serotonin released from dense granules upon immune-complex stimulation promotes platelet aggregation and vascular permeability through 5-HT receptors. 10 It also modulates cytokine production by innate and adaptive immune cells. 36 Reduced serotonin levels in SLE correlate with severe phenotypes and nephritis. 35
4.4. Calprotectin (S100A8/A9)
Another molecule that has prothrombotic activity is calprotectin (S100A8/A9). Calprotectin is a heterodimeric protein expressed by granulocytes, monocytes, and platelets in inflammatory conditions. One of its receptors on the surface of platelets is called GPIV (CD36), which participates in the process of thrombosis by binding to them. 50 The findings indicate an increase in calprotectin during SLE, which is related to the activity of cardiovascular disease (CVD).50,51 Calprotectin is elevated in many SLE cohorts, but not all studies correlate it strongly with cardiovascular events. Differences in flare timing, neutrophil activation, and ethnicity likely explain variations.50,52
4.5. Complement
Activated platelets support complement activation and subsequent deposition of its fragments on platelets by exposing several molecules such as phosphatidylserine and chondroitin sulfate and binding to C1q. 53 Interestingly, the amount of complement C4d fragment, which is a product of C4, increases in patients with SLE, which can be deposited on the surface of other hematopoietic cells, including RBCs and platelets. Studies indicate that the increase of platelet C4d (PC4d) has a positive relationship with higher risk of thrombosis in the future for these patients, so that PC4d > 13 mean fluorescence intensity (MFI) is a predictor of vascular thrombosis with high sensitivity and specificity. 54 This will be further explained in the next section.
4.6. IFN I
IFN I is another factor causing thrombosis in patients with SLE. High serum levels of this cytokine in SLE are associated with a decrease in the number of endothelial progenitor cells and impairment in the ability of these cells to differentiate into mature cells, which can significantly increase the risk of atherosclerosis. Also, life-long exposure to IFN I in SLE can affect the transcriptomes and proteomes of megakaryocytes by increasing the expression of proteins regulated by IFN I (CD69-CD58-IFITM1-PRKRA) on the surface of these cells and subsequently the platelets produced from them. 32 Interestingly, these platelets are more active and remarkably increase the risk of venous thrombosis in these patients. There is a significant correlation between the levels of proteins regulated by IFN I in patients with SLE with a history of vascular disease (VD) compared to patients without a history of VD, which can be another mechanism to explain vascular disorders and thrombosis in patients with SLE. 32
4.7. Neutrophil extracellular traps (NETs)
NETs provide a link between inflammation and thrombosis. In fact, the process of NETs provides a network for adhesion, activation, and aggregation of platelets and thus participates in thrombosis of SLE. 55
Thrombosis factors of platelets.


Platelet activation mechanisms and the potential impact of activated platelets on SLE Thrombosis (a): Platelets are activated by immune complexes (ICs) via the Fc receptor, FcγRIIA. Also, the complement protein C1q enhances platelet activation by immune complexes. Viruses primarily trigger platelet activation via TLRs. (b): Platelet-produced soluble mediators, including S100A8/A9 proteins (calprotectin), serotonin, and sCD40L, have been linked to the development of thrombosis in SLE. Platelets also generate IL-1β, which stimulates endothelial cells. Platelet-derived microparticles (PMP), released from the membrane of activated platelets upon stimulation contribute to SLE thrombosis through various mechanisms. High levels of circulating platelet-leukocyte aggregates, induced by CD40-CD40L interactions, activate adaptive immune responses, including antibody production. Figure 1 was created entirely by the authors using Microsoft PowerPoint (Microsoft Office 365). No copyrighted material was used, and no permissions were required.
5. Platelets interaction with immune cells in SLE
Activation of platelets amplifies their interaction with the immune system. Following activation, platelets directly engage with leukocytes through the binding of P-selectin (CD62) to glycoprotein ligand 1 (PSGL-1) (CD162) on the surface of leukocytes, forming a binding connection. This primary binding is further intensified through interactions with various receptors, such as the binding of GPIb to (CD11b–CD18) contingent on the type of leukocyte involved. This interaction leads to mutual activation and the enhancement of immune response. 56 The aggregation of platelets and leukocytes establishes a crucial link between inflammation and thrombosis, namely two pivotal processes in the context of SLE. 57
5.1. Innate immune cells
Interactions between platelets and innate immune cells yield diverse and intricate effects on these cells. The binding of activated platelets to neutrophils through the P-selectin–PSGL1 axis influences the neutrophil phenotype, elevating CD11b expression levels and oxidative burst. This interaction also induces an excessive production of NETs, forming a platform for thrombotic events and serving as a potential source of autoantigens. HMGB1, produced by activated platelets, undergoes oxidation in the body, further facilitating the process of NETs. 58
The interaction between platelets and PDC through the CD40L-CD40 axis increases the production of IFN-α by PDCs, a cytokine implicated in various symptoms associated with this autoimmune disorder. Studies demonstrate a heightened accumulation of platelet-PDC complexes in patients with active SLE. 8 Monocytes are another cell type in contact with activated platelets, which is facilitated through the expression of PSGL1 and binding to P-selectin on the platelets surface, as well as CD40L-CD40 interaction. This interaction directs the maturation of monocytes into activated DCs by upregulating the expression of CD80 and CD86. Furthermore, the formation of monocyte-platelet complexes accelerates TF production by active monocytes, which contributes to coagulation. 33
The complement system, a vital component of the immune system, plays a main role in the pathogenesis of inflammation and tissue damage in SLE, with the classical pathway activated by antigen-antibody complexes being the most crucial. 44 Platelets express certain complement components, including C3, C4, C1q, C1qR, and complement activation supports platelet activation. 23 In active patients with SLE, there is a decrease in C3 and C4 levels and an increase in complement products, including C4b, iC3b, and iC3dg. 44 The iC3b/C3 and iC3dg/C3 ratios are elevated in active patients with SLE. Among these molecules, C4d is a promising marker for evaluating disease activity because its delta change in disease activity is higher relative to C3 and C4 in a person with low and high disease activity. 59 It is worth mentioning that the level of CD4 in patients with lupus nephritis is higher than in those without nephritis, which can to some extent serve as an alternative to invasive biopsies for evaluating active nephritis. Elevated C4d deposition on platelets is associated with the presence of anti-cardiolipin antibodies, lupus anticoagulant, higher disease activity, and a greater frequency of thrombotic events in patients with SLE. 60 Complement fragments, including EC4d on erythroid cells, PC4d on platelets, and BC4 on B cells, which are known as cell-bound complement activation products (CB CAPs), are attached to cell surfaces in patients with SLE. The measurement of these fragments through flow cytometry is a sensitive index for diagnosing and monitoring patients with SLE. Increased PC4d levels are associated with an elevated risk of thrombosis, especially venous thrombosis in this disease. 44
Furthermore, the binding of these complement molecules to cell surfaces exacerbates dysfunction and inflammation of lupus. Studies indicate that Cd4 deposition on RBC surfaces decreases oxygen delivery to tissues, and that Cd3 binding to T-cells increases the production of cytokines such as IFN, IL4, and IL17.61,62 As the level of immunoglobulins bound to platelets reflects the presence of immune complexes in the bloodstream, it is anticipated that immunoglobulin level is inversely related to plasma levels of complement proteins. Investigations into the ratio of complement protein levels and immunoglobulin heavy chains in patients with SLE reveal a negative correlation between the complement C3 protein level with the gamma heavy chain and an inverse correlation between plasma level of complement C4 protein and the μ heavy chain. 12 This research implies that interactions between platelets and innate immune cells have various and complicated impacts on these cells, and that these interactions could be used as therapeutic targets for controlling innate immunity and thrombogenicity.
5.2. Adaptive immune cells
Platelet–immune cell interaction has surfaced as a potentially crucial yet underrecognized axis in the development of systemic lupus erythematosus (SLE). Multiple studies indicate that Platelet activation and attachment to lymphocytes exert an influence on T-cell phenotype. Accumulations of platelet-CD4+ T-cells result in an expanded compartment of T follicular helper (TFH) cells, germinal center expansion in lymph nodes, isotype switching, and elevated antibody titers, ultimately contributing to increased disease activity. 8 P-selectin, PSGL1 involvement in Treg cells leading to a reduction in FOXP3 disrupts the suppressive function of these cells, promoting the production of autoantibodies and increasing platelet activation. 63 Similarly, The binding of platelets to B cells through CD40L-CD40 interaction stimulates immunoglobulin production and antibody class switching. The CD40L-CD40 axis facilitates T-cell-dependent B cell responses. Studies in humans indicate elevated levels of B cell-platelet aggregations in patients with SLE, correlating with circulating levels of pre-switched memory B cells and immunoglobulins. 57 The highest percentage of lymphocytes attached to platelets is observed in patients with SLE with active disease, a positive Anti-ds DNA titer, and renal manifestations. This suggests that platelet-lymphocyte interactions can alter lymphocyte function, increasing antibody production, switching isotype, enhancing cell survival, and finally escalating disease activity. 64 While these findings support a plausible mechanism linking platelets to autoantibody generation, the evidence remains heterogeneous, and it is not yet clear whether platelet–B cell aggregates contribute independently to serologic relapse or organ damage.
Platelets produce the inflammatory mediator platelet-associated lectin, galactoside-binding, soluble 3 binding protein (LGALS3BP), which contributes to the inflammatory process. LGALS3BP serves as a ligand for galectin, promoting cell adhesion and activating pro-inflammatory pathways. Increased LGALS3BP levels in patients with SLE are associated with an elevated risk of thromboembolism. Patients with nephritis also exhibit higher LGALS3BP levels. This protein induces the release of inflammatory cytokines such as IL-8, IL-6, MCP-1, TNF-ɑ, and βIL-1 in monocytes and macrophages, intensifying myeloid inflammation. On the other hand, macrophages stimulated by LGALS3BP are reinforced in the presence of INF-a and produce more IL-6, which creates inflammation with greater intensity. 3
In addition, platelets may also affect myeloid inflammation. Parameters related to immune cell-platelets interactions, such as the neutrophil to lymphocyte ratio (NLR) and platelet to lymphocyte ratio (PLR), are useful in evaluating SLE disease activity. A meta-analysis demonstrates a significant relationship between increased NLR and PLR in patients with SLE compared to the control group, indicating disease activity (SLEDAI). Moreover, patients with SLE with kidney involvement show higher NLR levels, which serves as an indicator of kidney involvement in these patients. 65
Taken together, these results indicate that these two parameters, i.e., NLR and PLR, measure the underlying inflammation or immunological disturbances of SLE disease activity rather than platelet activation alone. These parameters need to be used in conjunction with clinical characteristics and disease-specific markers, as they are non-specific and might also be influenced by other diseases such as infection, medications, or other forms of inflammation.
Analysis of platelet protein contents in patients with SLE using liquid chromatography with mass spectrometry reveals a higher number of proteins, including immunoglobulins, complement proteins, autoantigens, and proteins binding to phosphatidylserine that are involved in degranulation irrespective of their size. Conversely, proteins in the cell skeleton, those binding to microtubules that maintain platelet structure, macrophage migration inhibitory factor (MIF), CXCR4 receptor, and Lipopolysaccharide-binding protein (LBP) are higher in the control group. 12 These findings suggest that understanding the role of platelets and their interaction with immune cells in SLE can unveil promising therapeutic strategies for this potentially severe autoimmune disease. Targeting the function or accumulation of specific immune cells may offer avenues to improve manifestations and inflammation in SLE.
6. Platelets and their laboratory indices: Indicators of disease activity
Early diagnosis and treatment of Systemic Lupus Erythematosus (SLE) are crucial for preventing irreversible organ damage. Although diagnosis traditionally relies on clinical assessment and serological markers, the heterogeneity of symptoms and alternating periods of flare and remission complicate disease monitoring. Platelet parameters have emerged as potentially valuable adjunctive indicators of SLE activity.
Platelet count and derived indices—such as Mean Platelet Volume (MPV), Platelet Distribution Width (PDW), Platelet-to-Lymphocyte Ratio (PLR), and measures of immature platelets—may hold prognostic significance. Thrombocytopenia (TCP), a common hematologic manifestation, correlates negatively with disease severity and may indicate increased risk of atherosclerosis and vascular complications. 66
There are conflicting results in consensus on the relationship between disease severity and MPV in SLE patients. Some studies suggest an increase in MPV with a positive correlation with SLE disease activity, 7 while others associate reduced MPV with disease severity. 67 The decrease in MPV may be a function of the increase in apoptosis mechanism, which exists during SLE. 12 Conflicting results regarding its relationship with SLE disease activity are likely caused by the dynamic nature of platelet activation and clearance in SLE, differences in disease stage, and methodological variances. A synthesis of available data suggests that MPV behaves as a stage-dependent marker: it may increase during early immune-driven activation but decrease in later phases dominated by clearance and marrow compensation, explaining the bidirectional findings.68–70 Thus, MPV should not be used as a stand-alone biomarker but rather interpreted within the broader clinical context.
PDW, another crucial laboratory indicator, reflects platelets size heterogeneity and has been associated with increasing SLE activity. Some studies propose this factor as a valid and cost-effective marker for clinically evaluating patients with SLE. 71 The association of this factor with disease activity could be attributed to changes in megakaryopoiesis and platelet activation caused by inflammation. However, it is not disease-specific.
Studies have also been conducted on the relationship between immature platelets and clinical parameters in SLE. Immature Platelet Fraction (IPF) and Absolute Immature Platelet Count (AIPC) have been introduced as useful parameters for platelets recovery. Since the response to steroids and the number of megakaryocytes is important indicators in thrombocytopenic patients and given the difficulty of bone marrow aspiration and biopsy, evaluating these two parameters can be helpful in this regard. The immature platelet fraction (IPF) indicates newly formed platelets in the bone marrow and is associated with the severity of SLE, while the absolute immature platelet count (AIPC) predicts the response to steroid therapy in thrombocytopenic lupus patients that is important for physicians as it prevents excessive immune system suppression and facilitates clinical decisions.72,73 It should be noted that these laboratory indicators are not specific to SLE and can be influenced by other factors. Therefore, clinical manifestations and other laboratory findings must be considered in their interpretation. These indices are promising for monitoring platelet recovery and predicting response to therapy, although further research is necessary to elucidate their diagnostic and prognostic utility in SLE.
7. Autoantibodies against platelets in SLE
SLE is characterized by a diverse array of autoantibodies, with approximately 180 known autoantibodies identified so far. These antibodies target various autologous antigens, including cytoplasmic proteins, cell membrane antigens, phospholipid-associated antigens, endothelial cells, nervous system antigens, plasma proteins, and nuclear components like DNA and histones. 74
Autoantibodies against platelets in SLE.
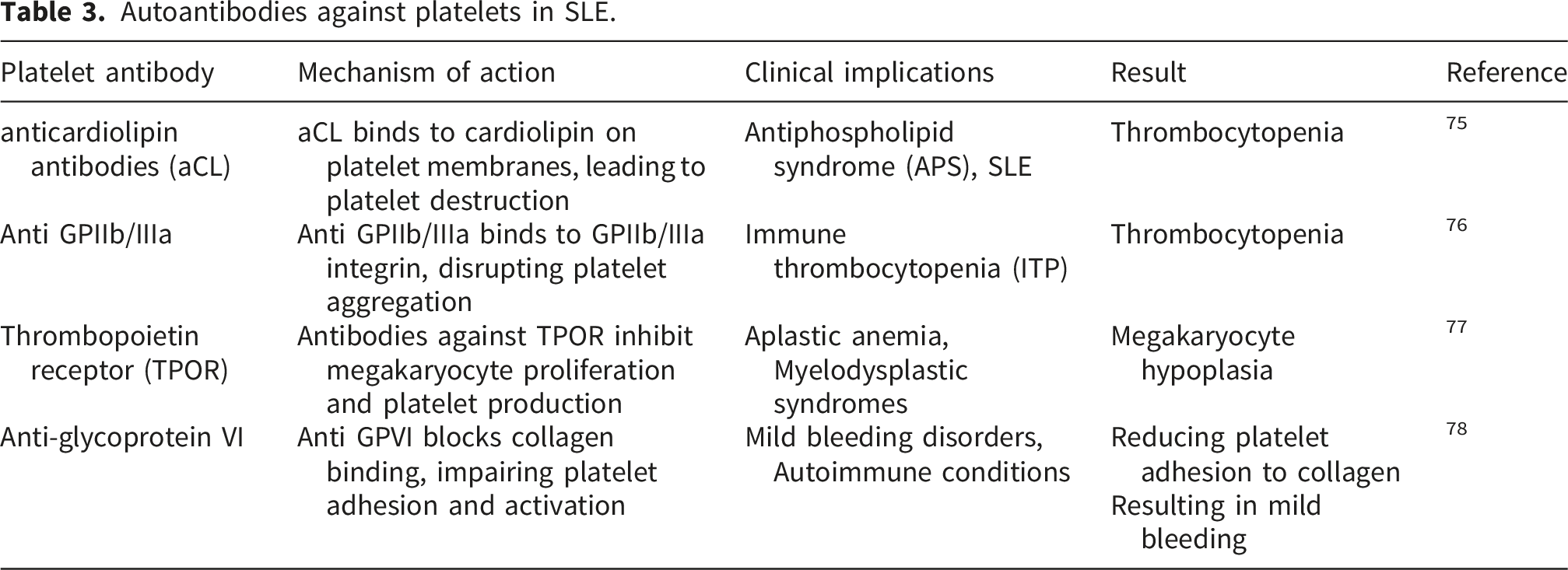
Recent studies also underscore the pathogenic role of autoantibodies targeting the GPIb/IX complex, a receptor central to vWF-mediated platelet adhesion and platelet clearance. Anti-GPIb/IX antibodies—well described in immune thrombocytopenia (ITP)—have been increasingly detected in autoimmune thrombocytopenias and may similarly contribute to SLE-related cytopenias. These antibodies can induce thrombocytopenia through multiple pathways, including Fc-mediated phagocytosis, platelet activation, and desialylation-driven clearance, mechanisms that simultaneously link immune destruction with heightened thrombotic risk. Experimental work in ITP shows that anti-GPIbα autoantibodies activate platelet signaling and promote FcγRIIa-dependent clearance, emphasizing their pathogenic relevance and potential applicability to SLE.79,80 The GPIb/IX axis is of particular interest because it is central to von Willebrand factor (vWF)–mediated platelet adhesion under high shear stress, initiating early platelet activation and contributing to thrombus formation. Detailed mechanistic studies have established that the GPIb-IX complex functions as the primary receptor for vWF and orchestrates downstream signaling events necessary for integrin activation and platelet aggregation. 81
Overall research on platelet autoantibodies in SLE is useful for understanding their role in the disease’s pathogenesis and clinical manifestations, including thrombocytopenia and vascular complications. These investigations provide valuable insights into disease activity. Moreover, platelet autoantibodies serve as potential biomarkers for predicting and monitoring thrombocytopenia and other complications in patients with SLE. This knowledge is essential for developing targeted therapeutic approaches to modulate platelet autoantibodies and improve patient outcomes.
7.1. Novelty statement
This review presents an integrated perspective on platelets as key mediators linking immune dysregulation, inflammation, and thrombosis in systemic lupus erythematosus (SLE). Unlike prior reviews that address these processes separately, it synthesizes emerging evidence on platelet activation pathways, platelet–immune cell interactions, platelet-derived microparticles, complement deposition, and type I interferon–driven platelet priming. By connecting mechanistic insights with clinical biomarkers and therapeutic implications, this work highlights platelets as underrecognized diagnostic and therapeutic targets in SLE.
8. Limitations
As a narrative review, this work may be influenced by publication bias and does not follow a systematic screening protocol. The available literature on platelet activation in SLE remains heterogeneous, with variation in methodology, patient populations, disease activity levels, and assays for platelet function or microparticle measurement. Moreover, many mechanistic insights are derived from small cohorts or experimental models, which may limit generalizability. These factors should be considered when interpreting the conclusions of this review.
9. Conclusion
Various mechanisms have been proposed to explain the pathophysiology of SLE. In this narrative review, we have summarized the current evidence for platelet activation in this disease. Further investigation has shown that platelets, a potentially critical player, may have a role in the pathogenesis of this disease, primarily through their dual involvement in inflammatory and thrombotic pathways. Although platelets are non-nucleated and are often overlooked as sources of nucleic acids and antigens in SLE, their mitochondrial content and mtDNA may support increased autoantigenic blood circulation and immune complex production in patients with SLE. Additionally, the detailed analysis of platelets and their derived PMPs, as well as their interaction with components of the innate and adaptive immune system, demonstrates platelets’ involvement in the thrombosis process, which is one of the main causes of mortality in patients with SLE. This study enhances our understanding of another factor involved in the pathophysiology of SLE. Potential platelet-based therapeutic and diagnostic approaches could include the development of drugs that specifically target platelet activation or inhibit the release of pro-inflammatory platelet mediators. Additionally, platelet-derived biomarkers, such as circulating PMPs, platelet surface receptors, or platelet-associated mtDNA, may serve as diagnostic or prognostic indicators, helping to identify patients at higher risk of thrombosis or disease flares and to monitor treatment response. Further studies should focus on validating the potential for the treatment of SLE and the phenotypic variations related to platelets. The inclusion of parameters related to platelets in the clinical evaluation of patients with SLE might improve predictions and treatment outcomes for the disease.
Footnotes
Author contributions
Study conception and design was performed by Najmaldin Saki, the first draft of the manuscript was written by Fatemeh Bakhshipour, manuscript revision was performed by Najmaldin Saki and Fatemeh Bakhshipour. Mehdi Torabizadeh and Emad behboodi provided critical review of the manuscript. Alireza mohebbi conceived and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.