
Abstract
Immune adjustment has become a sepsis occurring in the development of an important mechanism that cannot be ignored. This article from the perspective of immune infiltration of pediatric sepsis screening markers, and promote the understanding of disease mechanisms. Bioinformatics integrated six data sets of pediatric sepsis by using the surrogate variable analysis package and then analyzed differentially expressed genes (DEGs), immune infiltration and weighted gene co-expression network analysis of characteristics (WGCNA) of immune infiltration between pediatric sepsis and the control. Common genes of WGCNA and DEGs were used to functional annotation, pathway enrichment analysis and protein-protein interaction network. Support vector machine (SVM), least absolute shrinkage and selection operator (LASSO) regression and multivariate logistic regression were used to confirm the key genes for the diagnosis of pediatric sepsis. Receiver operating characteristic (ROC) curve, C index, principal component analysis (PCA) and GiViTi calibration band were used to evaluate the diagnostic performance of key genes. Decision curve analysis (DCA) was used to evaluate the clinical application value of key genes. Lastly, the correlation between key genes and immune cells was analyze. NK cells Resting and NK cell activated in pediatric sepsis during immune infiltration were significantly lower than those in the control group, while M1 Macrophages were higher than those in the control group. ROC, C-index, PCA, GiViTi calibration band and DCA indicated that MCEMP1, CD177, MMP8 and OLFM4 had high diagnostic performance for pediatric sepsis. There is a negative correlation between 4 key genes and NK cells resting, NK cells activated. Except for MCEMP1, the other 3 genes were positively correlated with M1 Macrophages. This study revealed differences in immune responses in pediatric sepsis and identified four key genes as potential biomarkers. Pediatric sepsis in pathology maybe understood better by learning about how it develops.
Keywords
Introduction
Pediatric sepsis is defined as a clinical syndrome characterized by the isolation of pathogens from normally sterile body fluids of children or the presentation of systemic inflammatory response syndrome (SIRS) induced by proinflammatory factors caused by severe pathogen infection, which is one of the major causes of death in children.1–3 Early identification of sepsis in children and aggressive treatment within hours of patients with sepsis may prevent irreversible progression and adverse outcomes.4,5
At present, the key to the diagnosis of sepsis still lies in the judgment of infection site and the identification of pathogenic factors. Blood culture is a traditional method, but it is the “gold standard” for the diagnosis of sepsis in children. Because blood culture is affected by external factors, such as the use of antibiotics will significantly affect the results of blood culture, and the positive rate of blood culture is positively correlated with the amount of blood collected.6,7 Similarly, the 16sRNA detection method contributes greatly to the diagnosis of sepsis, but the samples are prone to false positive detection due to external contamination. 8 Infection marker is an important way to diagnose sepsis in children. C-reactive protein (CRP) is an acute phase protein secreted by liver cells that can stimulate CRP secretion during microbial invasion and inflammatory reactions. 9 CD64 is the type I receptor of the Fc fragment of immunoglobulin G. 10 Usually, CD64 is hardly expressed on the surface of mature neutrophils, but it is expressed in large quantities in bacterial infection, and the level can be increased within 4–6 h. 11 The level of procalcitonin (PCT) in healthy people’s serum is extremely low. When bacterial infection occurs, toxins and inflammatory factors in the body can inhibit the degradation of PCT, resulting in an increase in PCT levels. 12 Therefore, CRP, CD64 and PCT have important reference value in the diagnosis of childhood sepsis.13–16 Interleukin-6 (IL-6) and interleukin-27 (IL-27), as pleiotropic cytokines, have both anti-inflammatory and proinflammatory effects and are key factors in the inflammatory mediator network. 17 Some studies have confirmed that the levels of IL-6 and IL-27 in sepsis patients are significantly higher than those in patients with systemic inflammatory response syndrome caused by noninfection, which can be used to differentiate them.17–20 Cytokines of interleukin-6 and interleukin-27 increased more rapidly than PCT in the early stage of inflammation, which had important clinical significance in the early diagnosis of sepsis in children and could evaluate the prognosis.19,21,22 Tumor necrosis factor-alpha (TNF-α), as a cytokine involved in systemic inflammation, is a member of the cytokine group when acute reactions are stimulated. 23 TNF-a has high sensitivity to predict neonatal sepsis, but has no specificity to identify bacterial infection and cannot be prognostic.24,25 However, children have unique pathophysiological characteristics, and some of the early symptoms of children are not typical, and the corresponding biomarkers and other examination results are difficult to provide clear diagnostic evidence in the first time. Therefore, it is particularly important to find sensitive early diagnostic indicators clinically and realize early intervention for children with sepsis.
In recent years, emerging molecular diagnostic technologies have been gradually applied in the diagnosis of sepsis in children,22,26–30 but they are still in their infancy. Further research on emerging technologies and continuous development need to be carried out to obtain faster and more accurate diagnosis. Gene chip technology is the reverse transcription of RNA into cDNA, and then the labelled cDNA (target) is applied to the surface of the support with the nucleotide sequence corresponding to the probe. The probe and the target are hybridized by standard nucleic acid interactions, and the number of hybridizations reflects the abundance of specific mRNA species. 31 The genomic changes of childhood sepsis were described by using gene chip technology, but comprehensive analysis results of various childhood sepsis chip data are still lacking. In this study, multiple pediatric sepsis gene expression chip data from the Gene Expression Omnibus (GEO) database were used for comprehensive analysis by integrated bioinformatics methods to identify common differential genes and conduct functional analysis. Meanwhile, diagnostic markers and immune cell infiltration characteristics of pediatric sepsis were explored. Finally, the findings of this study may be helpful to further understand the pathogenesis and development of sepsis in children, and may provide new methods for its diagnosis, prevention and treatment.
Materials and methods
Microarray data
Information on the included microarray data sets.

Identification of differentially expressed genes
Using “limma” package of six sepsis data set were analyzed, and to correct the adjust after p < 0.05 and | logFC | > 2 as standard to screen differentially expressed genes (DEGs), and use the “pheatmap” pack drawing heat maps of DEGs. 35
Immune cell infiltration analysis
In order to measure the relative proportion of infiltrated immune cells in sepsis, CIBERSORT (https://cibersortx.stanford.edu/), a method for analyzing immune cell types in different tissues, was used to analyze the combined expression data and calculate the infiltration of immune cells, and the proportion of 22 types of immune cells was set to run 100 times. 36 Suitable samples were screened according to p < 0.05. Calculate the percentage of each immune cell type in the sample and display it as a bar graph. Heat maps of 22 immune cells were made using the “pheatmap” package. The violot software package was used to compare and visualize the level samples of 22 immune cells between sepsis and the control group. A corrlot software package was used to create a heat map, revealing correlations between 22 infiltrated immune cells. Principal component analysis (PCA) was used to evaluate whether sepsis and control could be distinguished in immune infiltration.
Weighted gene co-expression network analysis (WGCNA) construction and identification of modules
To explore the interaction between genes, we used WGCNA to explore genes associated with pediatric sepsis. 37 First, genes with more than 25% variation between samples in the integrated dataset were introduced into WGCNA. Secondly, in order to ensure the reliability of network construction results, the outlier samples are removed. Thirdly, Pick-soft-threshold function is used to calculate the adjacency of the Soft Threshold weight β obtained by co-expression similarity. Then, the adjacency relationship is transformed into topological overlap matrix (TOM), and the corresponding dissimilarity degree (1-Tom) is calculated. Fourthly, module detection is carried out by hierarchical clustering and dynamic tree cutting functions. To classify genes with similar expression profiles into gene modules, average linkage hierarchical clustering was carried out according to the tom-based dissimilarity measure, and the minimum size of the gene tree (genome) was 50 [19]. Fifth, for modules associated with clinical attributes, module membership (MM) and gene significance (GS) were calculated. Finally, the eigenstate network is visualized. The genetic information in the module was used for further analysis. Common genes (CG) are the intersection of DEGs and genes in the significant module screened from integrated data set.
Functional annotation and pathway enrichment analysis
The “clusterProfiler” package in R software was used to analyze the GO and KEGG pathways of common genes, 38 and p < 0.05 was considered as statistically significant difference.
Protein-protein interaction network construction
Using the interactive gene retrieval (STRING) database retrieval tool (Version11.0; www.string-db.org) construct protein-protein interaction (PPI) with the minimum interaction score ≥0.4 as the truncation value.39,40
Candidate diagnostic biomarker screening
The above CGs were used as the key genes for further screening the diagnosis of pediatric sepsis. A support vector machine (SVM) is a supervised machine learning algorithm that can be used for regression or classification, based on a training group that detects and associates them with labels. SVM recursive feature elimination (RFE) is a machine learning algorithm that requires training on subsets of elements of multiple categories to reduce size in order to search for optimal variables. Least absolute shrinkage and selection operator (LASSO) regression models are further used to select optimal variables through the use of penalty coefficients. LASSO logistic regression and SVM-RFE were used to select the most significant characteristic genes, and the intersection results of those two methods were further used to screen key genes by multivariate logistic regression.41,42 According to the results of logistic regression, the diagnosis model of pediatric sepsis was constructed by incorporating genes.
Correlation analysis between identified genes and infiltrating immune cells
This study evaluated the relationship between immune cells and key genes in R software by calculating Spearman rank correlation coefficient. For visualization processing with “ggplot2” software package, p < 0.05 was considered to be statistically significant.
Statistical analysis
R software (version 4.0.5; https://www.r-project.org/) was used to perform all statistical analyses. Continuous variables were expressed by comparing the mean, standard deviation, and difference between the two groups. T test was used for normally distributed variables and Mann-Whitney U tests were used for nonnormally distributed variables. The receiver operating characteristic (ROC) curve, C index, principal component analysis (PCA) and GiViTi calibration band and GiViTi calibration band were used to evaluate the characteristic genes of differentiation between pediatric sepsis and normal controls. 43 Considering the potential clinical application value of establishing the gene diagnosis model, the decision curve analysis (DCA) evaluates whether the decision based on this model is beneficial to pediatric sepsis. 43
Results
Removal of the batch effect
In this study, 6 GEO data sets (GSE4607, GSE8121, GSE9692, GSE26378, GSE26440 and GSE80496) on pediatric sepsis were included. The workflow of this study was shown in Figure 1. Due to different factors such as samples and testing platforms, there was a certain batch effect. To eliminate the batch effect of samples, SVA packages were used in this study. The combination of six genes resulted in a total of 13,031 genes. Before removing the batch effect of data set samples, the samples were batch clustered according to the top two principal components (PCs) of the non-standardized expression values (Figure 2(a)). In contrast, the principal component analysis (PCA) based on standardized expression showed that the batch effect of different data sets was obviously eliminated (Figure 2(b)). In other words, the results of this method show that the standardization of different data sets has successfully eliminated the batch effect. Study flow chart. Gene expression data set based on principal component analysis.

DEGs in pediatric sepsis
A total of 11 DEGs (MCEMP1, CD177, HP, MMP8, GPR84, RETN, ANXA3, IL1R2, LCN2, LTF and OLFM4) were screened according to the screening conditions after batch correction and standardization of the 6 data sets. All 11 genes were up-regulated (Figure 3). The genes were differentially expressed between pediatric sepsis and control.
Immune cell infiltration analysis
CIBERSORT was used to screen 452 blood samples from sepsis patients and normal controls (p < 0.05), resulting in 361 blood samples. We calculated the different immune cells in the blood of the control group and the children with sepsis (Figure 4). NK cells resting and NK cells activated in the control group were higher than those in sepsis patients (p < 0.05). However, M1 Macrophages were higher in sepsis patients than in control group (p = 0.003) (Figure 4(b)). There was a positive relationship between T cells CD4 memory activated and T cells CD8 (0.45). Neutrophils and Mast cells Activated were proportional (0.45). Mast cells activated was negatively proportional to T cells CD8 (−0.47) and NK cells RESTING (−0.46), respectively. Neutrophils was also negatively proportional to T cells CD8 (−0.45) (Figure 4(c)). The PCA results showed that although the boundary between the sepsis and the control groups overlapped, there was a certain degree of differentiation between the two groups (Figure 4(d)). The landscape of immune cell infiltration between pediatric sepsis and control.
The WGCNA co-expression network of pediatric sepsis
First of all, after batch correction and standardization of the combined 6 data sets, genes were sequenced according to the size of variance, and genes with the largest variance 25% (3258) were selected for WGCNA analysis (Figure 5). Second, the flashClust toolkit was applied for cluster analysis, and the threshold was set to 70. Then, no outlier samples were detected. Third, in order to determine the appropriate soft thresholds, the ‘pickSoftThreshold' function in the WGCNA software package was used to filter from the power parameter setting from 1 to 20. The results show that when power = 16 (Figure 5(a) and (b)), the scale-free topological model and the fitting curve in the plateau area are gentle and stable. In order to avoid the similarity between each gene, the minimum number of genes in each gene network module was 50. Then the flashhClust software package was used for dynamic clustering analysis. First, dynamic crosscutting was used to generate a tree (Figure 5(c)). We also calculated the characteristic genes for each module to cluster the modules in parallel, especially once the correlation exceeded 0.8, and the modules merged together. Modular characteristic gene (MB) in blue module (r = −0.45; P = 1 × 10−23) showed the highest correlation with other modules, and was significantly correlated with NK cells Resting (Figure 5(d)). Therefore, there were 616 blue module genes, and the intersection with the 11 DEGs obtained above. Eleven common genes (CGs) were obtained as markers for further analysis of sepsis diagnosis in this study (Figure 5(e)). Weighted gene co-expression network analysis in pediatric sepsis and Venn diagram of the 11 CGs.
Functional annotation and pathway enrichment analysis of the CGs
In the GO analysis of CGs, the BP enrichment term was neutrophil degranulation, neutrophil activation involved in immune response and defense response to bacterium; CC accumulated terms were specific granule, tertiary granule and vesicle lumen; MF enrichment items are serine-type endopeptidase activity, serine-type peptidase activity and serine hydrolase activity (Figure 6(a)). KEGG enrichment analysis showed that these genes were significantly enriched in the IL-17 signaling pathway, Hematopoietic cell lineage and Fluid shear stress and atherosclerosis (Figure 6(b)). Functional enrichment analysis, KEGG and PPI of the DEGs.
PPI network construction
In the PPI network constructed by 11 CGs, there were 11 nodes and 15 edges (Figure 6(c)).
Diagnostic biomarker selection and construction of the prediction model
In order to screen out key genes with potential for the diagnosis of pediatric sepsis, 11 common genes were initially identified by LASSO and SVM-RFE. A total of 5 genes were identified by LASSO (Figure 7(a) and (b)) and 11 genes were identified by SVM-RFE (Figure 7(c) and (d)). Five common genes were screened using these two methods for further screening by multivariate logistic regression (Figure 7(e) and (f)). Finally, four genes (MCEMP1, CD177, MMP8 and OLFM4) were identified as key genes in the diagnosis of pediatric sepsis (Figure 7(f)). Both AUC and C-index (>0.9) indicated that these four genes had good diagnostic value (Figure 8(a)). The purpose of the GiViTi calibration band is to reveal the relationship between the predicted and observed probabilities by fitting polynomial logic functions. When diagonal bisectors are used and the 95%CI region is less than 45°, the fitting degree of the prediction model is better. p > 0.05 of GiViTi calibration curve indicated that the prediction model fitted well (Figure 8(b)). PCA also intuitively showed that these four genes could distinguish sepsis patients from normal group (Figure 8(c)). It could be seen from the DCA curve that the red line remained above the gray line and black line from 0 to 1, which indicated that the decision based on the model of four genes may be beneficial to pediatric sepsis (Figure 8(d)). The clinical impact curve suggested that the predictive ability of this model had certain potential clinical application value (Figure 8(e)). Pediatric sepsis potential diagnostic markers screened by LASSO, SVM-RFE and multivariate logistic regression analysis. Evaluation of potential diagnostic biomarkers of pediatric sepsis.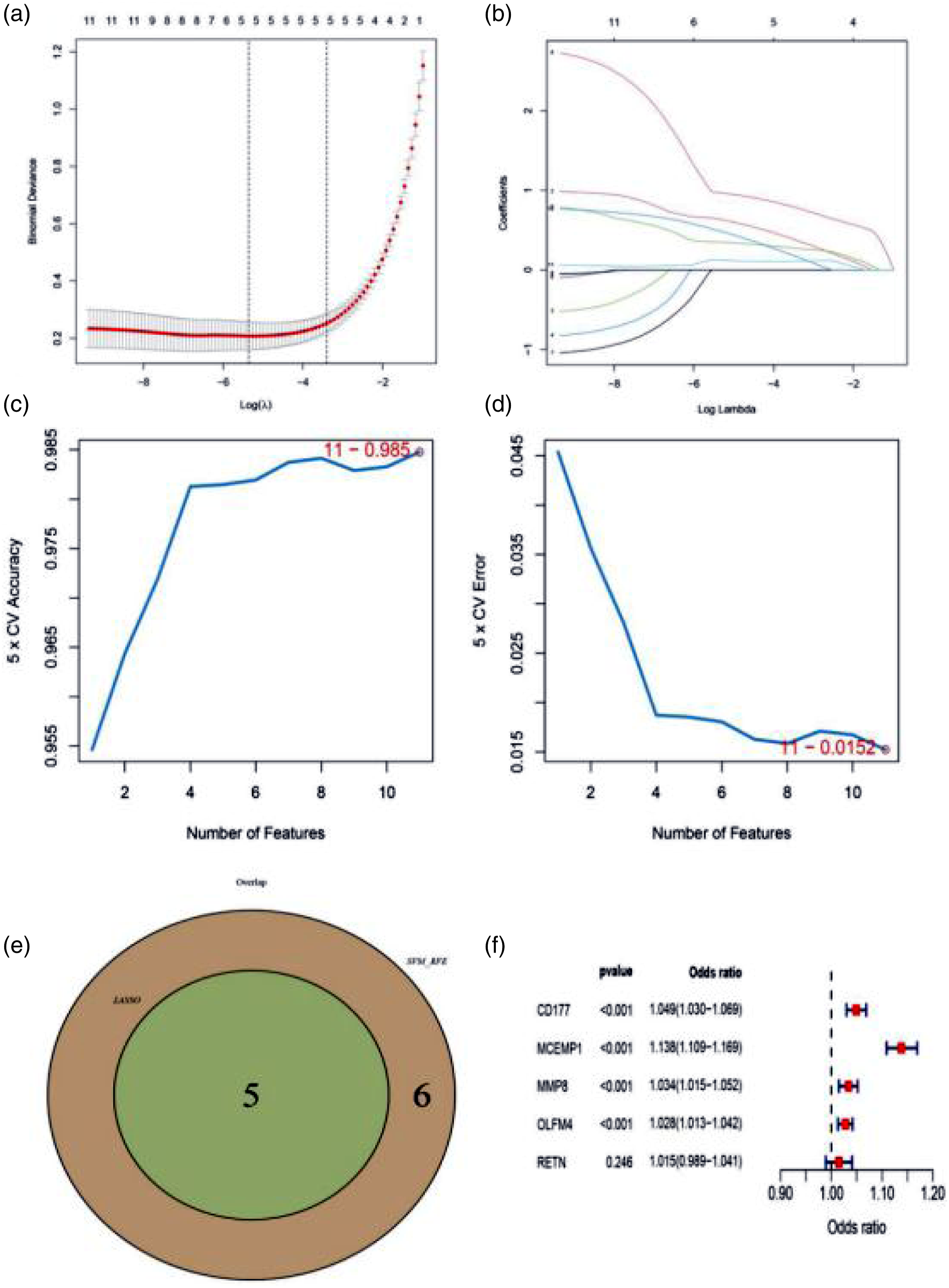

Correlation diagnostic biomarkers and infiltrating immune cells
Figure 9 shows the correlation between four diagnostic markers and immune infiltration. Among them, 4 genes (MCEMP1, CD177, MMP8 and OLFM4) were proportional to NK cell activated (p < 0.05). The correlation coefficient of CD177 with NK cells activated were 0.167, indicating that it was a highly correlated gene. Four genes were negatively proportional to NK cells RESTING (p < 0.05). Compared with NK cell activated, MCEMP1 had a high correlation with NK cells RESTING (correlation coefficient = −0.454). However, in M1 Macrophages, except MCEMP1, the other 3 genes were positively correlated with each other (p < 0.05). The correlation coefficient between CD177 and M1 macrophages was 0.218, which was the highest among the four genes (Figure 10). Correlation analysis of diagnostic biomarkers and immune infiltration in pediatric sepsis. Correlation analysis of diagnostic biomarkers and M1 Macrophages, NK cells activated and NK cells resting in pediatric sepsis.

Discussion
Sepsis is a severe inflammation of the body caused by infection. The pathophysiology of sepsis is characterized by the activation of a large number of inflammatory cells and the release of pro-inflammatory mediators in the early stage, and then the body enters into immunosuppression due to the depletion of inflammatory mediators and the apoptosis of immune cells. 44 Both proinflammatory and anti-inflammatory reactions occurred in the early stage of sepsis, and even mixed antagonistic reaction syndrome appeared. 45
Integration data sets
Because the clinical information of the data sets included in our study was incomplete, after integrating these six data sets, factors such as sex, age, race and prognosis of the sample source were not considered in the data analysis process of our study.22,26–30 However, the influence of the above factors on high-throughput data analysis was avoided. In this study, the SVA package is used to integrate data. The “SVA” package not only includes the ComBat function but is also used to remove batches and other unmeasurable or unmodeled variation sources. At the same time, it eliminates the batch effect. 34 It also has good compatibility with the “limma” package. The “limma” package was used to analyse the differential expression of microarray data. 35 The reason is that “limma” has now become the mainstream choice for gene discovery. This R package contains particularly powerful tools that can efficiently complete data reading and data exploration. Therefore, to compensate for some factors that have not been considered in the process of data analysis and affect the data results, the application of SVA packages will minimize the influencing factors. The reason is that according to experience, when there are a large number of known or unknown potential confounding factors, the adjustment of substitution variables may be more appropriate.
Immune infiltration of pediatric sepsis
In this study, 22 kinds of immune cells were analyzed in the immune infiltration of pediatric sepsis compared with the control group, M1 Macrophages, NK cells Resting and NK cells activated were significantly different (p < 0.05). Macrophages are multifunctional cells in the natural immune system, whose roles include host defense, cytotoxicity, clearance of apoptotic cells, and promotion of tissue repair. M1 macrophages play a key role in antigen presentation, proinflammatory cytokine secretion and phagocytic activity. It has been reported that a large number of M1 macrophages are expressed in mice with acute kidney injury caused by sepsis. 46 In this way, the reported results are consistent with M1 Macrophages in septic children in our study, which were significantly higher than those in the control group. 46 This suggests that the intervention of M1 Macrophages can start from the early activation of sepsis. It has been reported that hereinafter inhibits the polarization of M1-like macrophages, alleviates the inflammatory response, and provides a competitive therapeutic strategy for sepsis. 47 Suppressors of cytokine signaling serve as regulators of macrophage polarization and have a beneficial role in limiting inflammatory responses. In septic shock animal models lacking Suppressors of cytokine signaling (SOCS3), it has been found to bias macrophages toward the M1 phenotype, promoting inflammatory response. 48 In this study, the NK cells resting and NK cell activated in children with sepsis were significantly lower than those in the control group. Studies have shown that sepsis can lead to a decrease in the number of immune cells such as natural killer (NK) cells and a decrease in the secretion of interferon gamma, suggesting that sepsis can lead to immunosuppression. Immune suppression during sepsis leads to secondary infection, which is also an important cause of late death of patients with sepsis.49,50 These results indicate that NK cells are related to the occurrence and development of sepsis, further suggesting that the intervention of sepsis can also start from NK cells. 51 It has been reported that the harmful effects of NK cells during sepsis are mediated by their ability to amplify pro-inflammatory responses or directly lead to organ damage, possibly through cytotoxic mechanisms. Mice depleted of NK cells not only exhibited systemic inflammation and hypothermia, enhanced microbial clearance, restored acid-base balance, and improved survival in experimental models of multistin-induced sepsis.52,53
Immune infiltration related four genes
In this study, differential genes were screened from six data sets of pediatric sepsis, and then WGCNA analysis was performed on six data sets of pediatric sepsis with clinical features of immune infiltration. The common genes obtained by intersection of the module gene set and DEGs of sepsis-related immune infiltration in children are the key genes for the diagnosis of pediatric sepsis. Then, LASSO, SVM-RFE and multivariate logistic regression analysis were successively applied to the common genes of the above two genes. Genes related to immune infiltration were screened as potential biomarkers for the diagnosis of sepsis in children. Then, the diagnostic performance of biomarkers was evaluated by ROC curve, C index, PCA and GiViTi calibration bands. The clinical application value is demonstrated by the clinical work curve drawn by DCA. Finally, four genes were selected as the key genes for the diagnosis of sepsis in children. In the evaluation of the diagnostic performance of the four genes, it can be shown that the four genes screened in this study have certain diagnostic ability. At present, some studies have reported the relationship between these 4 genes (CD177, MCEMP1, MMP8 and OLFM4) and pediatric sepsis.
CD177 is a neutrophil specific molecule involved in neonatal alloimmune neutropenia and respiratory infections. 54 CD177 was significantly higher in the lung tissue than in the control group in a mouse model of sepsis induced by cecal ligation perforation (CLP), suggesting that CD177 may be a new biomarker of sepsis. 55 CD177 was found in clinical studies to be up-regulated in transcriptional and phenotypic expression of neutrophil CD177 in patients with septic shock compared with healthy volunteers. 56 CD177 combined with other genes (IL1R2, OLFM4 and RETN) were better than acute physiology and Chronic health evaluation (APACHE) and sequential organ failure in predicting survival of patients with sepsis Assessment (SOFA) is more accurate. 57
Mast cell expression membrane protein 1 (MCEMP1) gene is mainly expressed in monocytes and mast cell lines in mast cells, and is involved in the pathogenesis of allergic and inflammatory diseases. 58 In the establishment of cecal ligation puncture sepsis mouse model, the high expression of MCEMP1 can be detected, indicating the pathological mechanism of its involvement in sepsis. Down-regulation of lncRNA NEAT1 may promote the inhibitory effect of Mir-125 on immune function of sepsis mice by inhibiting MCEMP1, suggesting that MCEMP1 is a potential therapeutic target for sepsis. 59 Similarly, down-regulation of lncRNA MALAT1 can inhibit the inflammatory response of sepsis, and the mechanism involved is closely related to up-regulation of Mir-23a and down-regulation of MCEMP1. 60
Matrix metalloproteinase-8 (MMP-8), also known as neutrophil collagenase, is a zinc-dependent endopeptidase in the MMP family. It is mainly expressed and secreted by neutrophils as a proenzyme (pro-MMP-8) and activated by reactive oxygen species released by activated neutrophils, so MMP-8 plays a key role in both acute and chronic diseases.61–63 The expression level of MMP-8 in patients with sepsis is higher and proportional to the severity of patients, suggesting that MMP-8 may be involved in the host response of sepsis. 64 MMP-8 also showed the same superior ability in the diagnosis of early onset sepsis (EOS). 17 In a young model of multibacterial sepsis, MMP8 helped clear bacteria from the abdominal cavity, suggesting that MMP8 loss has a negative impact on survival. But adult mice did benefit from MMP8 inhibition. 65
Olfactomedin4 (OLFM4), a member of the coumarin-domain glycoprotein family, is stored in the granules of mature neutrophils and its expression is regulated by G-CSF, PU1 and NF-kb.66,67 Recent functional studies have shown that OLFM4 as an important regulator is not only related to neutrophil sterilization, but also closely related to host natural immune resistance to Gram-positive and Gram-negative bacteria.68,69 OLFM4 gene expression has previously been reported as a biomarker for the diagnosis of sepsis. 70 The expression level of OLFM4 in sepsis patients was higher than that in control group. 71 The study also reported that juvenile OLFM4 deficient mice were protected from sepsis and thus from death, 72 similarly, patients with a high proportion of OLFM4 + neutrophils had a higher burden of organ failure and a higher risk of death. 73
Limitations
This study also has some limitations. Firstly, because there are no clinical data in those data sets, the clinical information cannot be considered, and it was not possible to analyze the relationship between diagnostic genes and prognosis in pediatric sepsis. Second, The CIBERSORT algorithm only draws from limited genetic data, so data analysis of immune infiltration may deviate from cellular atypia, disease-induced disease, or phenotypic plasticity. Finally, the data and results of this study need to be future confirmed by later clinical studies.
Conclusions
In this study, bioinformatics methods were used to identify potential immune markers for the diagnosis of sepsis in children. Eleven common genes in DEGs and WCGNA identified four key genes (MCEMP1, CD177, MMP8 and OLFM4) that recognized the NK cells RESTING related modules. The calibration bands of AUC, C-index, PCA and GiViTi calibration curve showed that these four genes had high differentiating ability between the diagnosis of sepsis in children and the control group. Accordingly, these four genes could serve as potential markers for the diagnosis of sepsis among children. Moreover, DCA also showed that the model of these four genes was beneficial to pediatric sepsis. These findings will help to understand the pathogenesis of pediatric sepsis, but further studies are needed to support our conclusions.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We thanked the S&T Program of Chengde (grant nos. 202006A088 and 202006A049).
Ethics approval
Ethical approval for this study was obtained from the Medical Ethics Committee of The Affiliated Hospital of Chengde Medical University (APPROVAL NUMBER/LL202001).