
Abstract
Objective
Ischemia/reperfusion (IR)-induced myocardial arrhythmias are a common clinical manifestation in patients with myocardial infarction after reperfusion therapy. Mitochondria play a critical role in cardioprotection. Here, we investigated the effects of KH176 as a new mitochondrial-acting drug on IR-induced ventricular arrhythmias, mitochondrial function, pro-inflammatory cytokines production, and the role of mitochondrial ATP-dependent K (mK-ATP) channels in rats’ hearts.
Methods
The hearts of Sprague Dawley rats (250 ± 30 g; 36 rats) underwent 35 min of ischemia followed by 120 min of reperfusion. Myocardial in vivo ischemia was induced by ligation of the left anterior descending coronary artery. KH176 at concentrations of 10 and 50 μM was intraperitoneally injected to rats 10 min before reperfusion onset. Ventricular arrhythmias were quantified during reperfusion, and cardiac mitochondrial function, nitric oxide, and pro-inflammatory cytokines levels were measured by fluorometric, spectrophotometric, and ELISA techniques.
Results
Administration of KH176 significantly reduced lactate-dehydrogenase release and the number, duration, incidence, and severity of ventricular arrhythmias induced by reperfusion injury. IR-induced elevation of mitochondrial reactive oxygen species production, and cardiac pro-inflammatory cytokines TNF-α, IL-6, and IL-1β, as well as reduction of mitochondrial membrane potential, ATP and nitric oxide levels were significantly restored by KH176 at 50 μM. However, the blockade of mK-ATP channels by 5-hydroxydecanoate considerably inhibited the effects of KH176 on all parameters except nitric oxide.
Conclusion
KH176 showed strong cardiac antiarrhythmic effects on IR-induced injury through improving mitochondrial function and reducing inflammatory and oxidative responses, and these protective effects are mediated by cardiac mK-ATP channels.

Introduction
Myocardial ischemic disorders are one of the leading health problems worldwide, leading to disability and high mortality. 1 Ischemia and reperfusion (IR) injuries in ischemic heart disorders and following many therapeutic interventions to establish adequate perfusion after ischemia are imposed on the heart, which if left untreated can cause myocardial infarction (MI) and will have irreversible or mostly irreversible consequences. 2 Reperfusion-induced arrhythmias are a common clinical manifestation in patients with MI after reperfusion therapy. The arrhythmogenic consequences of reperfusion injury are numerous and include an imbalance in oxidative stress, exacerbation of inflammatory responses, disturbances in the production and propagation of cardiac impulses, microcirculation abnormalities, and impaired normal mitochondrial function.1,2
The maintenance of intracellular electrolyte homeostasis and cellular metabolism in cardiomyocytes depends on the physiological function of mitochondria. 3 Meanwhile, ATP-dependent potassium channels located in the mitochondrial membrane play a critical role in maintaining this cellular homeostasis and are considered an important therapeutic target for cardioprotection. 4 However, these channels are blocked during IR damage following the increase of free radicals and ion disruption, thus disrupting mitochondrial self-coordination and integrity and increasing the production of reactive oxygen species (ROS) and inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukins.4,5 All of these make the severity of IR heart damage worse. There is ample evidence that opening these channels during reperfusion inhibits IR-induced ventricular arrhythmias. 6 In addition, nitric oxide (NO) is located upstream of these channels, and increased NO production in IR injury setting may open these protective channels, leading to protection of the heart against fatal arrhythmias.4,6
KH176, also known as sonlicromanol, is a bio-available small molecule that is being tested in clinical trials and has strong antioxidant effects and enhances the physiological features of mitochondria in vital cells.7,8 Preliminary studies postulated that the beneficial effects of this new drug, which is currently being studied in clinical trials of different phases, are sufficient to protect the brain and heart against IR injury.7,9 In confirmation of this hypothesis, a recent study has shown that this drug increases prostaglandin E2 (PGE2) activity by acting on mitochondria and decreases MI outcomes by increasing its antioxidant power and decreasing inflammatory responses. 10 Therefore, considering the therapeutic potentials of this drug and also due to the involvement of mitochondrial dysfunction and inflammatory responses in the development and severity of ventricular arrhythmias following cardiac IR damage, in this study we decided to evaluate the effect of this drug on reperfusion-induced ventricular arrhythmias, cardiac mitochondrial function and pro-inflammatory cytokine production, and explore the role of mitochondrial ATP-dependent K-channels (mK-ATP) in these effects.
Materials and method
Animals
Thirty-six Sprague Dawley rats (250 ± 30 g) were prepared from the laboratory animals breeding center of the university and kept in standard conditions of the animal house with a temperature of 22 ± 2°C and a humidity of 55% in a 12-hour light-dark cycle. All stages of research and treatment of animals were carried out based on the ethical approval of the local Committee for Animal Ethics (TPHH-REC-2081).
Induction of cardiac IR injury
Following a 2-week adaptation period, the animals were anesthetized with pentobarbital sodium (50 mg/kg, intraperitoneally). After cutting the intercostal muscles between the fourth and fifth ribs and exposing the heart, the left anterior descending coronary artery was tied with 5-0 silk thread for 35 min. The paleness of the left ventricular surfaces indicated the success of coronary occlusion. Then, by releasing the suture, reperfusion was established for 2 h to accomplish cardiac IR injury modeling in rats. During the procedures, the animals were placed on a heating pad to prevent their body temperature from dropping.
Sample size calculation and animal grouping
According to the power calculation for estimation of sample size, considering a type I error of 0.05, an expected 20% difference in the mean of endpoints, 10% in standard deviation, and a power of 85%, six rats per group was calculated. Rats were randomly divided into the following six groups (six rats each):
(1) Control; (2) IR; (3) IR + KH10; (4) IR + KH50; (5) IR + 5HD; and (6) IR + 5HD + KH50. In the KH-receiving groups, the KH176 was intraperitoneally injected into rats at concentrations of 10 μM (KH10) and 50 μM (KH50), 10 min before establishing reperfusion. 10 The drug was prepared in 1% DMSO and the non-treated IR animals received the same volume of DMSO intraperitoneally. To inhibit mK-ATP channels, 5-Hydroxydecanoate (5HD) at a concentration of 5 μM was injected intraperitoneally 5 min before injection of KH176 at 50 μM.
Recording and interpretation of ventricular arrhythmias
Two golden recording electrodes and one neutral electrode were used for electrocardiography, using a data recording and acquisition system (Harvard apparatus, USA). Ventricular arrhythmias including premature ventricular complex (PVC), ventricular tachycardia (VT), and ventricular fibrillation (VF) were recorded during reperfusion, and their number, duration, and incidence were calculated and interpreted using the LAMBETH convention for animal arrhythmias (Figure 1). The severity of arrhythmias was calculated and scored based on the occurrence or non-occurrence of each arrhythmia in rats in each group so that the absence of arrhythmia received a score of zero, the occurrence of PVC received 1, VT 2, reversible VF 3, and irreversible VF received a score of 4. ECG recording representative of ventricular arrhythmias in rats. PVCs: premature ventricular complexes, PVB: premature ventricular beat, VT: ventricular tachycardia, and VF: ventricular fibrillation.
LDH measurement
To measure the release of lactate dehydrogenase (LDH) from cardiac cells, the blood samples from the tail veins of rats were collected in the 30th minute of the reperfusion phase and the content of this enzyme was measured using a special kit by spectrophotometry (MyBioSource, Inc., USA). The optical absorption of the resulting solution was read at 340 nm and normalized to U/l.
Measurement of mitochondrial function indices
First, fresh samples taken from the left ventricular ischemic region were homogenized in mitochondrial isolation buffer in the presence of protease inhibitor (Sigma-Aldrich, USA), and then centrifuged at 10,000 g. The resulting pellets were then suspended again in the isolation solution and centrifuged at 21,000 g. For detecting mitochondrial ROS levels, the resulting supernatant, which is the mitochondrial fraction, was incubated for 30 min at 37°C in a phosphate-buffered solution (PBS) containing 2 μmol DCFDA dye (Sigma-Aldrich, USA). Then, using a fluorimeter, the amount of excitation and its emission were measured at 480 and 530 nm, respectively. The obtained values were calibrated based on the protein concentration of the samples and reported as mitochondrial ROS levels. Another amount of mitochondrial supernatant was incubated in 2 mL of PBS containing 2 μL of JC-1 dye (Sigma-Aldrich, USA) for 30 min at 37°C in a dark environment. After washing with PBS, the fluorescence intensity of JC-1 in each sample at red and green wavelengths was measured by a fluorimeter and the red to green ratio was used as an indicator to assess the degree of mitochondrial membrane depolarization.
Measurement of ATP production
Determination of ATP production from the left ventricle was performed using the corresponding bioluminescence assay kit (Sigma-Aldrich, USA). Accordingly, 10 mg fresh samples were lysed in 100 μL of assay buffer. After adding the ATP probe to the solution, its absorbance was detected at 570 nm, spectrophotometrically. Then, the ATP levels were normalized and expressed as nmol/mg protein.
Measurement of pro-inflammatory cytokines
After obtaining samples from the left ventricular ischemic region at the end of reperfusion, the samples were homogenized in RIPA buffer solution and after centrifugation and preparation of their supernatant, the contents of pro-inflammatory cytokines were measured using specific ELISA kits according to kit instructions (MyBioSource, Inc., USA). The protein content of the samples was also determined by the Bradford technique. The concentration of cytokines in each sample was standardized and reported based on the protein concentration of the samples.
Measurement of NO metabolites
First, the supernatant of the ventricular samples was deproteinized with 15 mg/kg zinc sulfate, and then 100 μL of the supernatants and 100 μL of vanadium chloride were poured into each well of a microplate to convert all nitrates in the sample to nitrite. After incubation, 50 μL of 2% sulfonamide and 50 μL of 0.1% N-(1-Naphthyl)ethylenediamine (Sigma-Aldrich, USA) were added to the wells and incubated for 30 min at 37°C. In another series of samples, these steps were performed without adding vanadium chloride to obtain nitrate concentration. The optical absorption of the samples was read at 540 nm and the amounts of NO metabolites were calculated and expressed as nmol/mg of sample protein.
Statistical analysis
The data were reported as mean ± SD. After confirming the normal distribution of data, the differences between the groups were analyzed by one-way analysis of variance (ANOVA) and Tukey post hoc test. Arrhythmias count and numbers were analyzed with non-parametric Kruskal-Wallis test. The incidence of arrhythmias was analyzed using Chi-square test. The minimum level of significance was considered at the alpha level of 0.05.
Results
Reduction of IR-induced ventricular arrhythmias by KH176, and its reversal by 5HD
Following in vivo myocardial IR injury by 35 min ischemia and 120 min reperfusion in Sprague Dawley rats, the number of ventricular arrhythmias including PVC (212.5 ± 21.3 vs 16.5 ± 1.5), VT (25.5 ± 2.6 vs 3.3 ± 0.5), and VF (15.2 ± 1.5 vs 2.0 ± 0.6), the duration of PVC (240 ± 25.7 vs 17.3 ± 1.5 s), VT (66.8 ± 6.1 vs 6.5 ± 0.5 s), and VF (75.3 ± 7.5 vs 14.5 ± 1.5 s), the incidence of VT (100% vs 33%) and VF (100% vs 17%) as well as the severity (score) of arrhythmias (3.66 ± 0.5 vs 1.33 ± 0.8, p < .001) were significantly increased in comparison to control group experiencing no IR injury (Figures 2(a)–(d)). Administration of KH176 at 10 μM (KH10 group) significantly reduced only the timing of PVC (175.7 ± 18.1 vs 240 ± 25.7 s) and incidence of VT (67% vs 100%) and VF (50% vs 100%), but it had no impact on the number, timing, and severity of other arrhythmias. However, administration of KH176 at 50 μM (KH50) considerably and more effectively reduced all arrhythmias indices as compared with the IR group (Figures 2(a)–(d)). Blockade of mK-ATP channels by 5HD per se did not influence ventricular arrhythmias, but it significantly inhibited the effects of KH176 at 50 μM on the number, timing (p < .001), incidence (p < .05) and severity (p < .001) of ventricular arrhythmias as compared with IR + KH50 group (Figures 2(a)–(d)). (A–D). Reduction of reperfusion-induced ventricular arrhythmias by KH176, and its reversal by 5HD in rats. (n = 6), **p < .01 and ***p < .001 vs control group; +p < .05, ++p < .01 and +++p < .001 vs. IR group; #p < .05, ##p < .01 and ###p < .001 vs IR+KH50 group. PVC: premature ventricular complex, VT: ventricular tachycardia, VF: ventricular fibrillation, IR: ischemia-reperfusion, KH10 and KH50: KH176 at 10 m
Reduction of IR-induced LDH release by KH176, and its reversal by 5HD
LDH release was significantly elevated after myocardial IR injury, and administration of KH176 at 50 μM but not 10 μM significantly impeded IR-induced elevation in LDH release (p < .01; Figure 3). Notably, co-administration of 5HD significantly abolished the effect of KH176 on LDH release when compared with that of the KH50 group (p < .01). Reduction of IR-induced myocardial LDH release by KH176, and its reversal by 5HD in rats. (n = 6), ***p < .001 vs control group; ++p < .01 vs IR group; ##p < .01 vs IR+KH50 group. IR: ischemia-reperfusion, KH10 and KH50: KH176 at 10 m
Reduction of IR-induced mitochondrial dysfunction by KH176, and its reversal by 5HD
Mitochondrial ROS production, mitochondrial membrane potential, and cellular ATP changes were evaluated to assess the mitochondrial function of the myocardium. IR injury significantly increased the production of mitochondrial ROS and its membrane potential and reduced ATP levels as compared with the control group (p < .01; Figures 4(a)–(c)). Although KH176 at 10 μM tended to restore these parameters following IR injury, the changes were not statistically significant. However, the administration of KH176 at 50 μM significantly reversed IR-induced elevation of mitochondrial ROS and membrane potential (p < .01) and reduction of ATP levels (p < .5). Moreover, concomitant blockade of myocardial mK-ATP channels significantly diminished the protective impacts of KH50 on mitochondrial function parameters (p < .01 and p < .05; Figure 4). (A–C). Reduction of IR-induced cardiac mitochondrial dysfunction by KH176, and its reversal by 5HD in rats. (n = 6), **p < .01 vs control group; +p < .05 and ++p < .01 vs IR group; #p < .05 and ##p < .01 vs IR+KH50 group. IR: ischemia-reperfusion, KH10 and KH50: KH176 at 10 m
Reduction of IR-induced myocardial pro-inflammatory cytokines by KH176, and its reversal by 5HD
To appraise the inflammatory status of myocardial tissue, the myocardial contents of pro-inflammatory cytokines including TNF-α, IL-6, and IL-1β were quantified (Figure 5). The production of pro-inflammatory cytokines was significantly increased following IR injury (p < .01). KH176 at 10 μM only reduced the production of TNF-α significantly, as compared to the IR group (p < .05). Instead, this drug at 50 μM significantly reduced IR-induced elevation of TNF-α, IL-1β (p < .001), and IL-6 (p < .01), in comparison to the IR group. Again, the anti-inflammatory impact of KH176 on all myocardial cytokines was considerably reversed after co-administration of 5HD, as the blocker of mK-ATP channels (p < .05; Figures 5(a)–(c)). (A–C). Reduction of IR-induced myocardial pro-inflammatory cytokines by KH176, and its reversal by 5HD in rats. (n = 6), **p < .01 vs control group; +p < .05, ++p < .01 and +++p < .001 vs IR group; #p < .05 vs IR+KH50 group. IR: ischemia-reperfusion, KH10 and KH50: KH176 at 10 m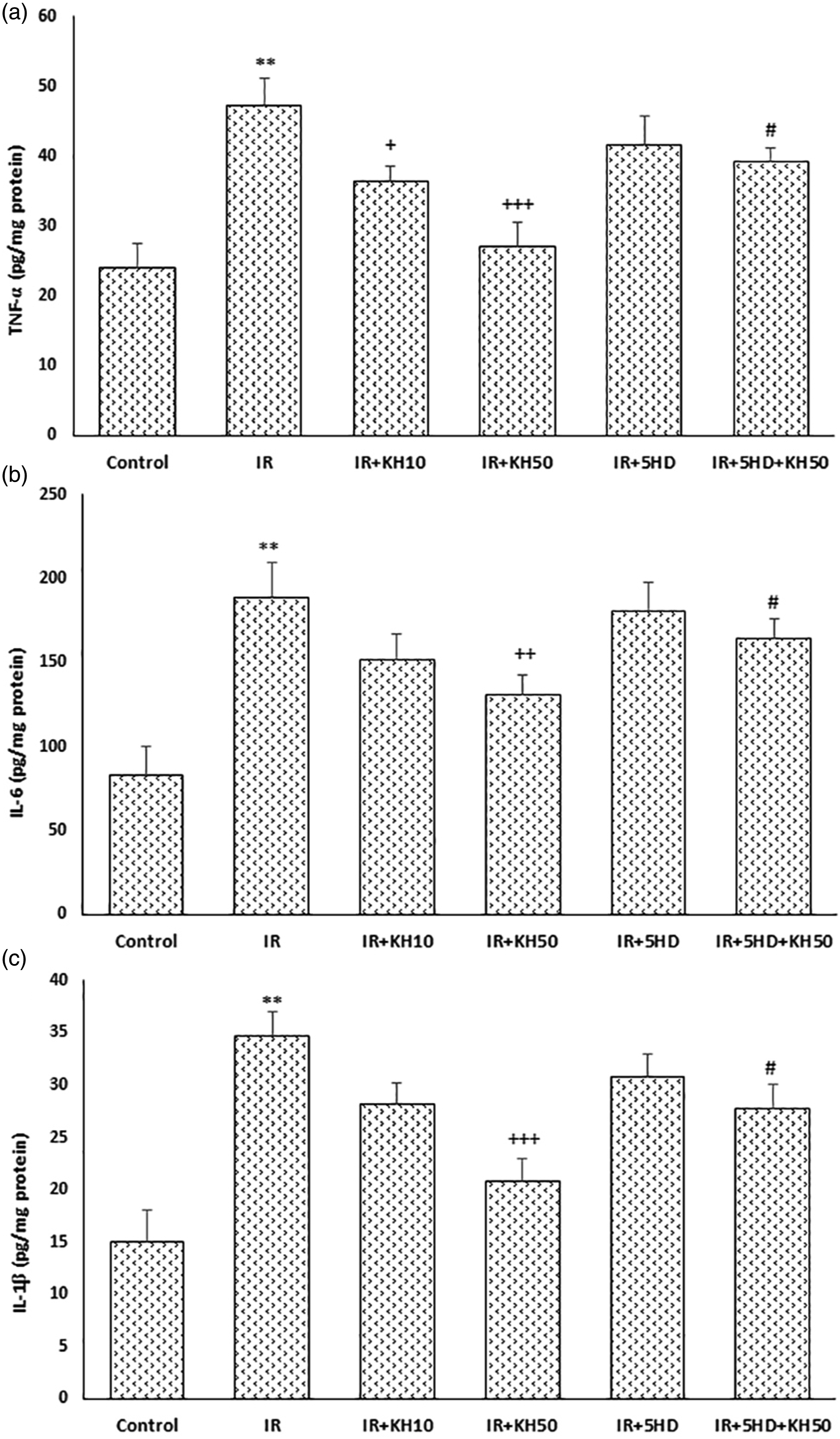
Elevation of IR-induced myocardial nitric oxide by KH176, which 5HD did not affect it
The myocardial content of NO, as assayed by NO metabolites, in the IR group was significantly lesser than in the control group (p < .001; Figure 6). KH176 at both dosages significantly upturned IR-induced reduction of NO levels (p < .05 for KH10 and p < .001 for KH50). Importantly, inhibition of myocardial mK-ATP channels using 5HD did not hinder the effect of KH176 therapy on NO production, as compared with the IR + KH50 group (Figure 6). Elevation of IR-induced myocardial nitric oxide by KH176, which 5HD did not affect it in rats. (n = 6), ***p < .01 vs control group; +p < .05 and +++p < .001 vs IR group. IR: ischemia-reperfusion, KH10 and KH50: KH176 at 10 m
Discussion
Cardiac arrhythmias are a major clinical consequence of reperfusion therapy, which can lead to increased mortality from IR damage by interfering with cardiac function. Therefore, the management of arrhythmias in the clinic and the modification of the underlying mechanisms are extremely important. In this study, the administration of the KH176 as a postconditioning modality was able to significantly diminish the occurrence and severity of ventricular arrhythmias and reduce the incidence of VT and VF to zero (at the concentration of 50 μM). The drug also increased NO production and decreased inflammatory cytokines production. Mitochondrial function was also improved and mitochondrial oxidative stress was inhibited by KH176 administration. Blocking mK-ATP channels by 5HD significantly eliminated the antiarrhythmic and anti-inflammatory effects of the drug and reduced its positive effect on cardiac mitochondria (Figure 7). Schematic diagram of the study. mK-ATP channels: mitochondrial ATP-sensitive K channels, NO: nitric oxide.
Due to their extensive function, mitochondria have a special contribution in protecting the heart against fatal arrhythmias. Following normal mitochondrial function, cellular oxygen is used for mitochondrial redox activity and ATP production, and the likelihood of high mitochondrial ROS production is reduced and the expression of antioxidant enzymes such as MnSOD is increased. 11 Otherwise, by beginning abrupt reperfusion, the defective mitochondria cannot consume much oxygen and inevitably increase free radicals, exacerbating IR circumstance by damaging cell membranes, proteins, and intracellular organelles. 12 Following disruption of mitochondrial function, the production of inflammatory cytokines in the cell also intensifies.11,12 Accordingly, increased oxidative stress and exacerbation of inflammatory responses are the two main arrhythmic mechanisms that occur in IR injury. 13 The use of KH176 in the present study prevented the production of pro-inflammatory cytokines and inhibited mitochondrial oxidative stress. Reduction of mitochondrial membrane depolarization following KH176 administration also indicates that this drug has been able to improve the electrolyte and ionic balance across the mitochondrial membranes and thus increase the stability of membrane potential. As might be expected, these changes will prevent ventricular arrhythmias.
KH176 is a new drug with a small molecule nature that has been reported to have strong antioxidant effects and according to initial observations, its main target is mitochondria and has a high potential in the modification and treatment of mitochondrial diseases.7,9 It can also prevent inflammatory responses by inhibiting the production of prostaglandin E2. 14 In line with our current results, the active metabolite of this drug has reduced IR-induced cardiac damages and corrected mitochondrial myopathy by modifying mitochondrial redox activity. 10 The protective effects of this drug have been dependent on the duration of ischemia. Previous studies have shown that this drug modifies the biological consequences of mitochondrial complex-1 dysfunction (seen in Leigh syndrome).15,16 Therefore, based on the findings of our study, which is consistent with the initial reports of the potentials of KH176 in mitochondrial-related diseases, this drug can modify the oxidative/phosphorylation activity of cardiac mitochondria in IR injury conditions. Similarly, balancing the mitochondrial membrane potential by the drug prevents mitochondrial dysfunction following reperfusion. Under these conditions, cellular ATP is produced to an optimal level and the generation of pro-inflammatory cytokines and the subsequent development of inflammatory responses are substantially inhibited. In the same way, the cellular milieu favors the better functioning of survival mediators to confine heart damage and reperfusion-induced lethal arrhythmias.
To further investigate the role of KH176 in controlling arrhythmias and inflammatory responses following IR injury, mK-ATP channels were inhibited by 5HD to evaluate the involvement of these channels in the protective effect of the drug. Inhibition of these channels not only eliminated the antiarrhythmic effects of the drug significantly but also reduced its effect on the production of inflammatory cytokines and mitochondrial function markers. It is inferred from these findings that the opening of these channels at the beginning of reperfusion mediates the cardioprotective effects of KH176. The mK-ATP opening has been reported to be accompanied by modification of ionic homeostasis across the mitochondrial membrane and prevention of mitochondrial swelling and membrane potential collapse. 17 These channels are considered as one of the main arms of mitochondria in playing their role as the final effector of cardioprotection against IR damage4,18 and therefore the drug has acted in reducing cardiac arrhythmias through this main pathway.
Finally, it is believed that NO can have protective impacts on the IR hearts by opening these channels. 19 In this study, although the drug increased the production of NO metabolites, inhibition of mK-ATP channels by 5HD could not reverse this effect, indicating that the effect of KH176 therapy on myocardial NO production in IR hearts was not dependent on the opening of mK-ATP channels. Furthermore, the effect of KH176 on NO levels in the presence of 5HD indicates that NO production in this situation occurs upstream of these channels. The association of NO changes with the antiarrhythmic effects of the drug indicates its potential protective effect on this phenomenon, but because these NO effects did not occur through the activation of mK-ATP channels, it seems that mediators or pathways other than mK-ATP channels are also involved to mediate the protective effects of NO. Further studies are suggested to understand the role of additional mediators and pathways such as PKG/cGMP and PI3K/AKT pathways and mitochondrial permeability transition pores. 20
Limitations
Changes in intramitochonrial or intracellular levels of electrolytes such as Na+, K+, and Ca2+ play an important role in regulating the electrical function of the heart and incidence of myocardial arrhythmias. But, their measurement was not as one of the objectives and hypothesis of this study. Increased oxidative stress as a result of mitochondrial dysfunction also raises the risk of myocardial arrhythmias, which was not examined in this study and requires further investigation.
Conclusion
KH176, as a new mitochondrial-acting drug, showed strong cardiac antiarrhythmic effects on IR-induced injury, that were associated with its effects on improving mitochondrial function and reducing inflammatory and oxidative responses, and these protective effects are mediated by increased activity of mK-ATP channels. Therefore, this drug can be considered as a better option to prevent the consequences of mitochondrial dysfunction in cardiac IR injury and the resultant arrhythmias. Additional studies are needed to clarify all aspects of the capacities of KH176 in this area, including its preconditioning rather than postconditioning effects.
Footnotes
Acknowledgements
We would like to express our special thanks of gratitude to the staff of Departments of Pharmacy and Cardiology at Jiangsu Taizhou People’s Hospital and Hubei Third People’s Hospital, China, for their helps and providing facility to this project.
Authors’ contributions
All authors designed the project, performed the experimentations, analyzed, and interpreted the data. GL was the major contributor in writing the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
All stages of research and treatment of animals were carried out based on the ethical approval of the local Committee for Animal Ethics (TPHH-REC-2081).
Availability of data and material
The authors confirm that the data supporting the findings of this study are available upon request.
Animal welfare
The present study followed international, and institutional guidelines for humane animal treatment and complied with relevant legislation.