
Abstract
Objective:
This study investigated whether and how intermedin (IMD) exerted a protective effect against simulated hypoxia/reoxygenation (H/R) injury in high-glucose-treated H9c2 cells.
Methods:
Cellular viability was assessed via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. Oxidative stress was determined by malondialdehyde and superoxide dismutase content in the culture medium supernatant. Flow cytometry with Annexin V/propidium iodide staining was used to detect the cardiomyocyte apoptosis rate. The protein expression of Bax, Bcl-2, caspase-3, and ERK1/2 was determined by western blot.
Results:
IMD administration to H9c2 cells during H/R injury decreased oxidative stress product generation and inhibited apoptosis (P < 0.05 or P < 0.01) while these effects were blocked by the ERK1/2 inhibitor (P < 0.05 or P < 0.01). Through the application of a specific ERK1/2 inhibitor, it was demonstrated that IMD mitigates high-glucose-induced oxidative stress and apoptosis via ERK1/2 signaling.
Conclusion:
Intermedin may be a novel therapeutic agent for mitigating diabetic cardiovascular injury in the clinical setting.
Introduction
Cardiovascular complications account for significant morbidity and mortality in the diabetic population. 1 The comprehensive mechanistic understanding of pathways responsible for exacerbated cardiomyopathy and cellular apoptosis observed in diabetic conditions remains elusive.2–5 Hence, there is an urgent need for therapeutic agents efficacious against diabetic ischemic injury in the clinical setting.
Intermedin (IMD), also known as adrenomedullin 2 (ADM2), belongs to the calcitonin gene-related peptide (CGRP) superfamily. Peptide fragments (IMD1-47, IMD8-47, and IMD1-53) are generated from pre-proIMD by proteolytic cleavage. 6 Among these three fragments, IMD1-53 (referred to as IMD in this study) exhibits the most potent biological cardiovascular effect, 7 particularly in the pathologic processes that involve the circulatory and renal systems, 8 as well as congestive heart failure. 9 IMD augments cardiac contractility, 10 inhibits collagen synthesis, mitigates cardiac fibroblast proliferation, 11 and attenuates cardiomyocyte hypertrophy. 12
Recent studies have demonstrated that streptozotocin (STZ)-induced diabetes mellitus significantly exacerbates myocardial ischemia/reperfusion (MI/R) injury, blunting the protective effect of various therapeutic agents.13,14 Myocardial oxidative stress contributes to diabetic pathophysiology. Furthermore, hyperglycemia enhances oxidative stress and degrades the efficacy of antioxidant defenses, 2 while others have shown that IMD reduces MI/R-induced injury.7,15 IMD and receptor activity-modifying protein expressions significantly decreased in the adipose tissue of obese mice; reduced blood glucose, fasting serum insulin, and free fatty acid levels; improved glucose tolerance and insulin sensitivity; and increased the glucose infusion rate during a hyperinsulinemic-euglycemic clamp test, indicating ameliorated high-fat diet (HFD)-induced insulin resistance. 16 IMD is involved in obesity and its related metabolic disorders.17–19 IMD has been demonstrated to protect human macrovascular, microvascular, and cardiac non-vascular cells against I/R injury via AM(1)-receptor signaling. 8 Furthermore, IMD1-53 exerts potent cardioprotective effects against acute rat ischemic injury. 15 IMD(1-53) exerts cardioprotective effect against myocardial I/R injury through the activation of the Akt/GSK-3-beta signaling pathway to inhibit mitochondria-mediated myocardial apoptosis. 20 Previous studies from our group demonstrated that IMD decreased in the plasma of diabetic rats linked IMD with diabetes. 21 IMD reduced insulin resistance in HFD-induced obese mice through elevating thermogenesis in brown adipose tissue. 16
We have previously demonstrated that IMD administration reduces hyperglycemia-exacerbated MI/R injury via the reduction in oxidative stress, apoptosis, and inflammation in a diabetic rat model. 21 However, the specific molecular mechanism by which IMD exerts its anti-ischemia/reperfusion effect remains unknown.
Therefore, the aims of this study are (1) to determine whether IMD exerts a protective effect in the diabetic model of ischemia/reperfusion and (2) elucidate the responsible underlying mechanism for such effect.
Methods
Cell culture and hypoxia/reoxygenation
All experimental procedures were approved by the Animal Care and Ethics Committee of Shanxi Medical University. The cardiac cell line H9c2 was derived from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured and maintained in monolayer at 37°C in a humidified incubator with 5% CO2 in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum (FBS), 100 U/mL of penicillin, and 100 μg/mL of streptomycin. Cells were incubated with normal (5.5 mmol/L) or high (33 mmol/L) glucose concentrations (Sigma, St. Louis, MO, USA). Following exposure to normal-glucose or high-glucose medium for 72 h for the induction of H/R injury, the hypoxic conditions were obtained by a three-GasB incubator containing 94% N2 and 5% CO2. Cardiomyocytes were subjected to hypoxia/reoxygenation (H/R) by hypoxia for 2 h followed by 4 h of reoxygenation in all H/R groups.22,23 Control cells were incubated under normoxic conditions for equivalent durations in high-glucose DMEM. The medium was replaced every 2–3 days. Cells were expanded to new culture plates upon reaching 80% confluence.
Experimental groups
H9c2 cells were randomly divided into five groups and were subjected to the following treatments: (1) normal culture medium (control group); (2) high-glucose medium (33 mmol/L; HG group, Sigma); (3) high-glucose medium and H/R (SI/R group); (4) high-glucose medium, hypoxia/reperfusion, and IMD treatment (1 μM, dissolved in normal saline IMD group); and (5) high-glucose medium, hypoxia/reperfusion, IMD, and PD98059 (MAPK/ERK kinase inhibitor, final concentration of 20 μmol/L; PD group, St. Louis, MO, USA).24,25 After 48 h of treatment, cells were harvested for western blot analysis and the quantification of apoptosis and oxidative stress. All the experiments were repeated four times (n = 4; Figure 1).

Experimental protocol overview.
In order to assess cellular viability, MTT (0.5 mg/mL; Sigma–Aldrich, St. Louis, MO, USA, 20 µL/mL) assay was performed. Cells were plated in 96-well plates (Corning Inc., NY, USA) at a cellular density of 1 × 104 per well. Oxidative stress was measured via malondialdehyde (MDA) and superoxide dismutase (SOD) quantification. Cellular necrosis was determined via lactate dehydrogenase (LDH) assay. For these assays, cells were cultured at 1 × 106 cells per 25 cm2 culture flask (Corning Inc.).
Cellular viability assay
Cell viability was assessed via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma–Aldrich) assay. Briefly, at experiment conclusion, the supernatant of each well was removed and replaced with 100 μL of MTT (50 μg/mL) solution and incubated at 37°C for 4 h. Formazan salt crystals were dissolved in 50 μL of dimethyl sulfoxide. The plates were analyzed via an ELISA plate reader (Bio-Rad, CA, USA) at 490 nm.26,27 Cellular viability was defined relative to control

Cellular injury assay
Cellular injury was determined by measuring LDH activity as a function of LDH release into the cell culture medium according to manufacturer’s protocol (Sigma). Briefly, LDH activity was determined by measuring the increase in NADH absorbance at 450 nm at 25°C via spectrophotometry (UV-4802; Unico, NJ, USA). 28
Oxidative stress assay
Lysis buffer (RIPA buffer, 150 mM of NaCl, 1% N-40, 0.5% deoxycholate, 0.1% sodium dodecylsulfate, 50 mM of Tris-hydrochloric acid, 2 mM of phenylmethylsulfonyl fluoride and proteinase inhibitor cocktail, and pH 7.4) was employed to collect cell samples. MDA level (nmol/g protein) was detected using a commercial thiobarbituric acid-reactive substance assay (Jiancheng, Nanjing, China) according to manufacturer’s protocol.
Total SOD activity was measured via UV-4802 mode spectrophotometry (Unico) at 550 nm. The supernatant was assessed via the xanthine oxidase-cytochrome C method. One unit of SOD was defined as the amount of enzyme required to inhibit 50% of the rate of cytochrome c reduction at 25°C, which is expressed as units/mL.
Apoptosis detection by flow cytometry
Apoptosis was determined using a commercial Annexin V-FITC apoptosis detection kit (BD Pharmingen, CA, USA) according to manufacturer’s protocol. Briefly, 1×105 cells were collected and washed two times with cold phosphate-buffered saline. Then, 5 mL of Annexin V and 5 mL of propidium iodide (PI) were added to these cells, and re-suspended in 500 mL of ×1 binding buffer. These cells were gently vortexed and incubated for 15 min at room temperature in the dark. Flow cytometric analysis was performed. Annexin V labeled with fluorophore identified cells in early stage apoptosis. PI, a fluorescent nucleic acid binding dye, stained cells at the medium and late stages of apoptosis. The apoptotic rate of a particular gate region was defined as follows: apoptotic rate = (number of Annexin V-positive + number of PI-negative cells) / (total number of cells).
Western blot analysis
Total protein was extracted from cells using lysis buffer containing 50 mM of Tris-HCl (pH 7.5), 100 mM of NaCl, 5 mM of ethylenediaminetetraacetic acid, 1% Triton X-100 (v/v), 1 mM of sodium fluoride (NaF), 1 mM of Na3VO4, and 4 mg mL−1 of complete protease inhibitor cocktail. Protein content was determined via Lowry method using a DC protein assay kit (Bio-Rad). Then, 30 mg of protein was resolved on 10% or 12% gradient gels, and transferred onto the nitrocellulose membrane (Schleicher & Schuell, Keene, NH, USA). After blocking with 5% nonfat milk, the blots were probed with various antibodies overnight at 4°C. After washing, antibodies cleaved against caspase-3 (1:1000 dilution; Sigma), Bcl-2, Bax, pERK1/2 (1:1000 dilution, p44/42 threonine and tyrosine; Sigma), and β-actin (1:2000, Shenggong, China), respectively, overnight with agitation at room temperature.
After final washing, the blots were incubated with Super-Signal West Pico chemiluminescence substrate reagent (Pierce Biotechnology, Inc., IL, USA) and developed by Kodak Biomax film. The developed blots were scanned. Band intensities were determined by Quantity One Image Analysis Software (Bio-Rad). Results were expressed as the percentage of control.
Statistical analysis
Statistical analyses were performed using SPSS 10.0 (Windows). The results of all experiments were expressed as mean ± standard error of the mean (SEM), which was obtained from replicate treatments. Data were analyzed using analysis of variance (ANOVA), followed by Dunn’s post hoc test for comparison. A P-value < 0.05 was considered statistically significant. The significance level of this study was set as two-tailed.
Results
High-glucose treatment decreases, while IMD increases cellular viability during SI/R, an effect blocked by ERK 1/2 inhibitor
Cellular viability was assessed via MTT assay (Figure 2). High-glucose treatment decreases cardiomyocyte viability, which was further decreased after SI/R. IMD administration during SI/R significantly increases cellular viability compared to SI/R alone (P < 0.05). The administration of ERK1/2 inhibitor PD98059 during SI/R resulted in significantly decreased cellular viability compared to the IMD group (P < 0.05).

Cellular viability (via MTT assay; n = 4).
High-glucose treatment augments LDH activity during SI/R, while IMD inhibits LDH activity during SI/R, an effect blocked by ERK1/2 inhibitor
Cellular injury was assessed via LDH activity assay (Figure 3). The high-glucose medium (HG) group released significantly more LDH into the supernatant compared to controls (HG: 837.85 ± 41.34 U/L vs 531.54 ± 40.23 U/L, P < 0.05). SI/R further augmented LDH activity compared to high-glucose treatment alone (SI/R: 1619.28 ± 35.05 U/L vs HG 837.85 ± 41.34 U/L, P < 0.05). IMD administration attenuated LDH activity compared to the SI/R group (IMD: 1149.67 ± 43.35 U/L vs SI/R alone 1619.28 ± 35.05 U/L, P < 0.05). Administration of ERK1/2 inhibitor PD98059 during SI/R resulted in significantly increased LDH activity compared to high-glucose treatment alone (PD98059: 1672.08 ± 51.02 U/L vs HG 837.85 ± 41.34 U/L, P < 0.05) and resulted in LDH activity compared to SI/R treatment alone (P > 0.05).

Cellular injury assay (via measurement of LDH release into the culture medium; n = 4).
High-glucose treatments exacerbate oxidative stress during SI/R, while IMD attenuates oxidative stress during SI/R, an effect blocked by ERK 1/2 inhibitor
Assays measuring oxidative stress (SOD and MDA quantification assays) are presented in Figure 3. Reactive oxygen species (ROS) exacerbate lipid peroxidation, inducing tissue breakdown, which release MDA and adversely interferes with cellular metabolism. Among the body’s physiologic antioxidant defense mechanisms is the radical scavenger SOD, which is consumed in its neutralization of oxidative stress. Therefore, MDA and SOD content indirectly reflect oxidative stress. Compared to controls, high-glucose medium increased MDA concentration and decreased SOD activity in the supernatant (MDA: 7.63 ± 0.47 nmol/mL, SOD: 41.44 ± 1.09 U/mL, P < 0.05). After SI/R, MDA content significantly increased (30.41 ± 0.93 nmol/mL, P < 0.05), and SOD activity significantly decreased (25.52 ± 1.0 U/mL, P < 0.05). IMD administration significantly decreased MDA concentrations and increased SOD activity, compared to high-glucose treatment alone (MDA: 17.11 ± 0.72 nmol/mL, SOD: 36.24 ± 1.11 U/mL, P < 0.05). The administration of the ERK1/2 inhibitor increased MDA concentrations and decreased SOD activity, compared to the IMD group (MDA: 29.89 ± 0.83 nmol/mL, SOD: 24.61 ± 0.54 U/mL, P < 0.05). However, there was no statistical difference compared to SI/R treatment alone (P > 0.05) (Figure 4).
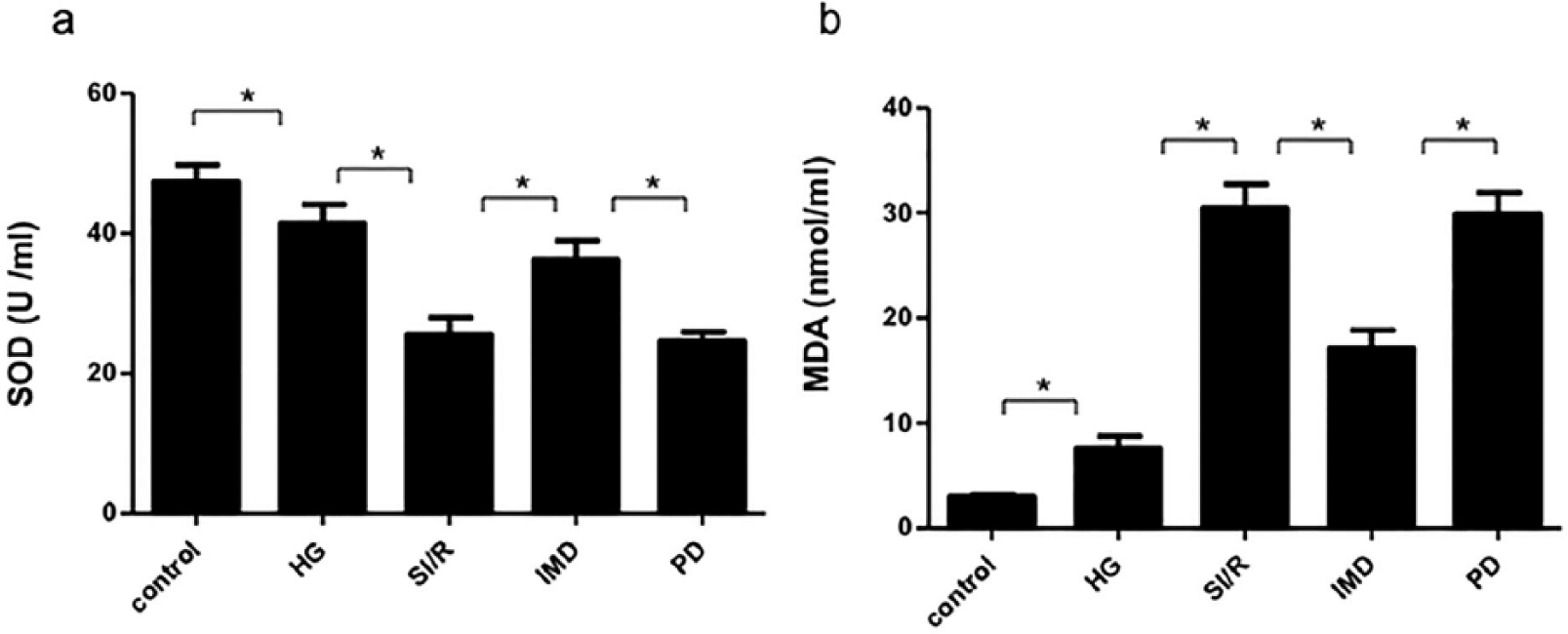
Oxidative stress assay (n = 4) via (a) SOD content and (b) MDA levels.
High-glucose treatments exacerbate apoptosis during SI/R, while IMD attenuates apoptosis during SI/R, an effect blocked by the ERK1/2 inhibitor
Flow cytometric assessment of apoptosis via Annexin V/PI staining is shown in Figure 4. High-glucose treatment significantly increased apoptotic rate in cells compared to controls (9.73 ± 0.15, P < 0.05). SI/R exacerbated apoptosis (65.47 ± 0.56), which was an effect mitigated by IMD administration (35.27 ± 0.72, P < 0.05). However, the effect of IMD was blocked by the administration of the ERK1/2 inhibitor (64.73 ± 0.21, P < 0.05), but there was no statistical difference compared to SI/R treatment alone (P > 0.05) (Figure 5).
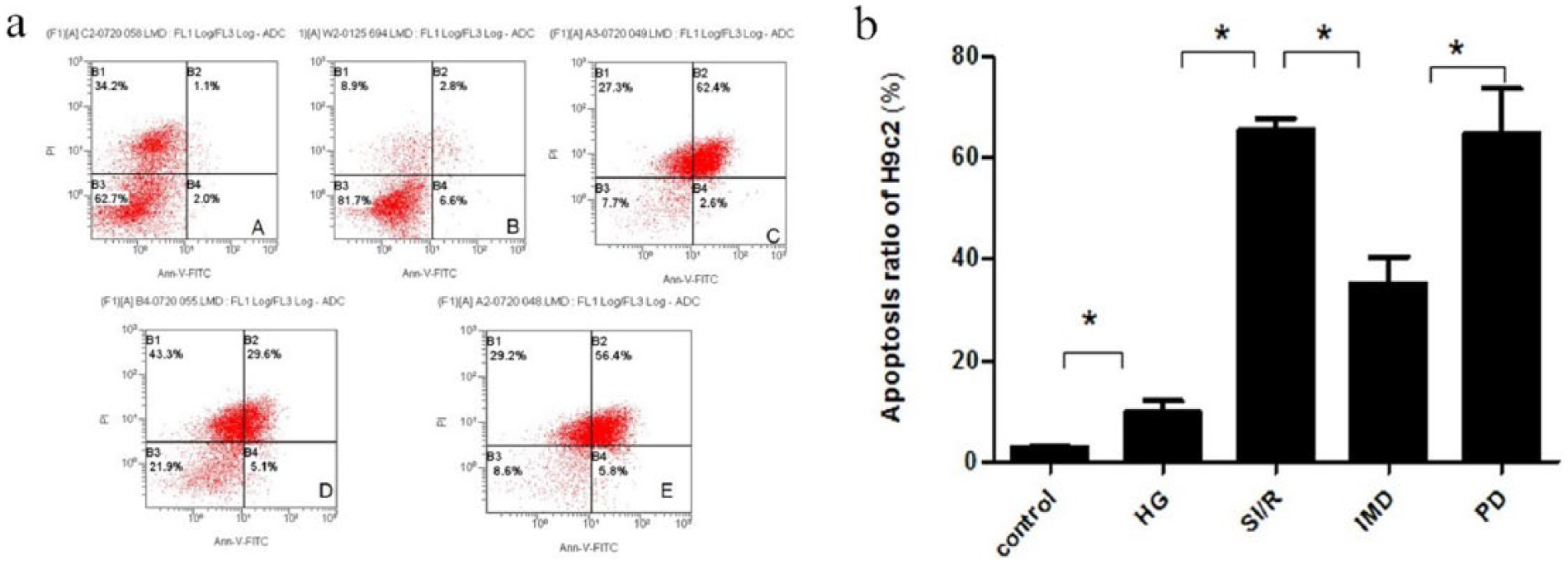
(a) Cardiomyocyte apoptotic rate was determined by flow cytometry with Annexin V/propidium iodide staining. (b) Apoptotic rate in the HG group was significantly greater than that in the control group. The apoptotic rate in the IMD group significantly decreased compared to the HG group (n = 6).
High-glucose treatments augment the expression of apoptotic proteins during SI/R, while IMD attenuates such protein expression, an effect blocked by the ERK 1/2 inhibitor
Western blot analyses demonstrated that after incubation with high-glucose, cardiomyocytes exhibited significantly increased apoptosis, increased expression of pro-apoptotic Bax and caspase-3, and decreased expression of anti-apoptotic Bcl-2 (Figure 6(a)–(d)). SI/R exacerbated apoptosis, an effect mitigated by IMD administration, which decreased caspase-3 activity, decreased Bax protein expression, and increased Bcl-2 protein expression (P < 0.05, Figure 6(a)–(d)). pERK1/2 protein expression significantly increased in the HG group compared to controls (P < 0.05, Figure 6(e)). SI/R increased pERK1/2 protein expression compared to HG group (P < 0.05, Figure 6(e)). IMD administration significantly decreased pERK1/2 protein expression compared to the SI/R group, an effect abrogated by the administration of ERK1/2 inhibitor PD98059 (P < 0.05, Figure 6(e)).

Western blot analysis, demonstrating expression of apoptosis-related proteins and pERK1/2 among various groups (n = 4). (a) Western blot analysis of activated caspase-3, Bcl-2, Bax, and PERK1/2 protein in H9c2 cells. Quantitative analysis of (b) Bcl-2, (c) BAX, (d) caspase-3, and (e) pERK1/2 western blots.
Discussion
High-glucose exposure induces cellular damage and apoptosis.29,30 Rats subjected to diabetic conditions manifested decreased cardiac function, increased oxidative stress, augmented apoptosis, and increased inflammatory response. ROS exacerbate lipid peroxidation, inducing tissue breakdown, and release MDA, which adversely interferes with cellular metabolism. The body’s antioxidant defense mechanism includes the radical scavenger SOD, which is consumed in its neutralization of oxidative stress. Therefore, MDA and SOD content indirectly reflect oxidative stress. This study demonstrates that high-glucose treatment exacerbates oxidative stress, increases LDH release, and decreases viability of H9c2 cells.
Previous studies7,15 support IMD as a promising cardiovascular protective molecule. It is protective against ischemia-reperfusion injury, improves cardiac function, mitigates cardiac hypertrophy, dilates blood vessels, and prevents vascular calcification.
MDA is an unsaturated fatty acid in free radical and lipid peroxidation metabolites. As an indirect marker of cellular damage degree, MDA content reflects the extent of systemic lipid peroxidation. The antioxidant SOD protects cells by reducing free radical-induced injury. SOD levels reflect the body’s capacity to scavenge oxygen-free radicals. In this study, myocardial SOD activity was attenuated in the HG group, which was further decreased by SI/R. In combination with the increased MDA content observed in the SI/R group, our data suggest that hyperglycemia-enhanced oxidative stress exacerbates SI/R injury. We confirm that high-glucose treatment (mimicking the diabetic condition) exacerbates membrane damage (increased LDH activity and MDA content), decreases antioxidant capacity (decreased SOD activity), and augments the apoptotic rate. IMD administration can effectively block the above effects. The potential responsible mechanism includes the following: (1) inhibition of cell membrane lipid peroxidation, thereby reducing injury and maintaining normal cellular structure and function; (2) significantly augment oxygen radical scavenger SOD activity, thereby maintaining balance between the physiologic antioxidant system and increased radical burden from high-glucose treatment; and (3) direct reduction in cellular apoptosis.
Merely found in eukaryotes, intracellular MAPK (mitogen-activated protein kinase) and serine/threonine protein kinase are involved in directing cellular responses to a diverse array of stimuli, and regulate proliferation, gene expression, differentiation, cell survival, and apoptosis. The three major MAPK signaling pathways include ERK (extracellular signal-regulated kinase), p38 MAPK (p38 mitogen-activated protein kinase), and SAPK JNK (c-Jun NH2-terminal kinase/stress-activated protein kinase) pathways. Diabetes and hyperglycemia significantly activate two pathways, the ERK and p38 MAPK pathways.31–33 The pathway discovered by others and the most comprehensively studied pathway is the ERK1/2 pathway, in which high glucose can obviously pass through the DAG-PKC activity ERK1/2, that is, high-glucose-DAG-PKC-ERK1/2.34–36 High-glucose-induced ERK1/2 phosphorylation has been implicated in the long-term deleterious effects of high glucose on β-cell, that is, apoptosis and impaired insulin secretory function. 37 Wolf’s study shows that high glucose stimulates ERK1/2, which phosphorylate p27Kip1 at serine 178, increasing its expression. 38 In our model, there is a need to investigate whether IMD can affect ERK1/2. Our results show that after applying inhibitors PD98059, IMD’s protective effect decreased, indicating that IMD exerts its protective effect through the ERK1/2 pathway. High glucose level and free fatty acids stimulate the de novo diacylglycerol (DAG)-PKC pathway, and subsequently stimulate ROS production through the PKC-dependent activation of NAD(P)H oxidase. 39 It has been demonstrated that antioxidants restore high-glucose-induced decreased DGK activity, mitigating the high-glucose-induced activation of the DAG-PKC pathway. 40 The activation of the diacylglycerol-protein kinase C (DAG-PKC) cascade has been implicated in the cardioprotective effects occurred after ischemia/reperfusion (I/R). Data have shown that high glucose can activate DAG and DAG-stimulated PKC, and the levels of phosphorylation activity of PKC further activated ERK1/2.41,42 As such, we specifically investigated the involvement of the ERK signaling pathway, employing the specific ERK1/2 inhibitor PD98059, which prevents ERK1/2 phosphorylation and its subsequent activation, ultimately leading to increased apoptosis.
In conclusion, we demonstrated that IMD administration may attenuate the augmented apoptosis associated with high-glucose treatments in an ERK1/2-dependent manner. IMD may represent a novel therapeutic agent for mitigating diabetic cardiovascular injury in the clinical setting.
Footnotes
Acknowledgements
The authors are particularly grateful to all the people who have given them help on their article. Hong Li, Chuan-Shi Xiao, Yun-Fei Bian, and Rui Bai acquired data. Hong Li, Chuan-Shi Xiao, Yun-Fei Bian, and Rui Bai drafted the manuscript. All authors contributed substantially to its revision. All authors take responsibility for the paper as a whole. All authors read and approved the final manuscript.
Availability of data and material
The authors declared that materials described in the manuscript, including all relevant raw data, will be freely available to any scientist wishing to use them for non-commercial purposes, without breaching participant confidentiality.
Declaration of conflicting interests
This study was conducted in accordance with the Declaration of Helsinki.
Ethical approval
The authors confirm that they have read the Editorial Policy pages. This study was conducted with approval from the Ethics Committee of the hospital.
Funding
This work was supported by a grant from the Shanxi Medical University, the second Hospital of Shanxi Medical University, and the Shanxi Provincial Health Department for Scientific and Technological Projects (no. 20100106).