
Abstract
Mitochondrial dysfunction is involved in the process of sepsis and leads to the accumulation of reactive oxygen species (ROS), which breaks cellular homeostasis and activates the downstream inflammatory cascade. The autophagic removal of ROS is a well-established cellular adaptive mechanism. Adiponectin is an adipocytokine that plays an important role in metabolic and inflammatory regulation. In this study, we investigated the anti-inflammatory effect of adiponectin in a sepsis model and its potential association with autophagy. We induced RAW 264.7 macrophages with lipopolysaccharide (LPS) to set up the sepsis model and treated them with adiponectin, an inhibitor of the nucleotide-binding domain and leucine-rich repeat containing family pyrin domain–containing 3 (NLRP3), ROS, Complex I, and an autophagy inhibitor. Flow cytometry and western blot analysis were performed to detect the expression levels of ROS, NLRP3, interleukin-1 beta (IL-1β), microtubule-associated protein 1A/1B-light chain 3II/I (LC3II/I), and adenosine monophosphate–activated protein kinase (AMPK). Expression levels of NLRP3, IL-1β, and ROS were significantly increased following LPS induction, and adiponectin reversed this up-regulation. Meanwhile, adiponectin also enhanced the expression of LC3II/I, an autophagosome marker, but an autophagy inhibitor and AMPK inhibitor depleted (reversed) the anti-inflammatory and antioxidant effect of adiponectin. Taken together, in the LPS-induced sepsis model, adiponectin alleviated the inflammatory reaction by reducing ROS production, possibly by enhancing autophagy via the AMPK pathway. The activation of autophagy may therefore be a key mechanism by which adiponectin ameliorates the inflammatory reactions of sepsis.
Introduction
Sepsis, the systemic inflammatory reaction caused by infection, is a major cause of mortality and morbidity. 1 The current treatment consists mainly of maintaining tissue perfusion and controlling infection. However, the result is usually poor. Several pathophysiological pathways are involved in sepsis. The pivotal factors in these processes are the accumulation of reactive oxygen species (ROS) and a consequent disruption of cellular homeostasis, resulting in oxidative stress and mitochondrial dysfunction. 2 Recent research shows that the nucleotide-binding domain and leucine-rich repeat containing family pyrin domain–containing 3 (NLRP3) play an important role in a vast number of diseases.3,4 The activation of NLRP3 requires mitochondrial ROS, and once activated, it activates the downstream inflammatory cascade. Furthermore, it aggravates related symptoms by amplifying inflammatory factors. 5 Mitochondrial dysfunction is also thought to be responsible for NLRP3 activation. Several lines of evidence indicate that mitochondrial impairment increases the mitochondrial DNA (mtDNA) copy number and mtDNA and subsequently contributes to the secretion of interleukin-1 beta (IL-1β) and IL-18 by acting through NLRP3. 6 Both ROS and mtDNA promote NLRP3 activation. 7 From this point of view, decreasing the level of ROS and protecting mitochondrial function are viable approaches to sepsis treatment.
The autophagic removal of damaged mitochondria is a well-established cellular adaptive mechanism to prevent cell damage. Researchers show that autophagy is also induced to sustain the organism during the process of sepsis. Autophagy may contribute to the reduction of oxidative damage by reducing the levels of oxidized substances. 8 Since autophagy is expected to decrease the level of ROS and since a lower ROS level is expected to suppress the inflammatory response, we hypothesize that facilitating autophagy might relieve the inflammatory response in sepsis.
Adiponectin (APN) is an adipocytokine released by adipose tissue and plays an important role in metabolic regulation and inflammation. 9 Previous studies demonstrate that APN protects against cardiac remodeling by suppressing inflammation through the activation of macrophage autophagy via the adenosine monophosphate–activated protein kinase (AMPK) pathway. 10 A study with a high-fat diet found that APN stimulated skeletal muscle autophagy and the antioxidant potential to reduce insulin resistance. 11 Research on acute liver injury also revealed that APN provides a barrier against mitochondrial dysfunction by promoting autophagy. 12 The above-mentioned studies suggest that the anti-inflammatory role of APN is associated with autophagy promotion, while it has not been reported whether APN decreases the level of oxidative stress in the process of sepsis by promoting mitochondrial autophagy.
In this study, we aimed to investigate the expression of inflammatory and autophagy-associated indices in lipopolysaccharide (LPS)-activated macrophages. We also examined whether the anti-inflammatory effect of APN was associated with an enhancement of autophagy via the AMPK pathway. We revealed that the activation of autophagy is a key mechanism by which APN might contribute to treating sepsis.
Materials and methods
Antibodies and reagents
Anti-LC3 (microtubule-associated protein 1 light chain 3), anti-NLRP3, anti-AMPK, glibenclamide, (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate (APDC), rotenone, compound C, and 3-methyladenine (3-MA) were obtained from Cell Signaling Technology (Boston, MA, USA). Recombinant globular APN was purchased from Sigma-Aldrich (St. Louis, MO, USA). Rabbit polyclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was obtained from SAB Signalway Antibody (Nanjing, China).
Cell culture
RAW 264.7 murine macrophages were purchased from the American Type Culture Collection. The cells were maintained in Dulbecco’s modified Eagles’ medium (DMEM) supplemented with 10% (v/v) fetal calf serum and 1% penicillin (Invitrogen, Carlsbad, CA, USA) at 37°C in a 5% CO2 incubator.
Cell experimental design
APN suppresses the activation of NLRP3 induced by LPS
Cells cultured in regular medium were incubated with LPS (500 ng/mL) for 24 h to establish the LPS group, and APN (30 μg/mL) alone was used for 30 min to establish the APN group. APN (30 μg/mL) was applied for 30 min before LPS treatment to establish the lipoic acid (LA) group. Glibenclamide (50 μM) was applied for 30 min before LPS treatment to set up LG50 group.
APN regulates NLRP3 activation via the modulation of ROS production
LPS (500 ng/mL) was applied for 24 h to establish the LPS group, and APN (30 μg/mL) was applied for 30 min before LPS treatment to establish the LA group. The ROS inhibitor APDC (50 μM) was applied for 30 min before treatment with APN followed by LPS to establish the LAA group, and the Complex I inhibitor rotenone (5 μM) was applied for 30 min before treatment with APN followed by LPS to establish the LAR group. ROS production was determined by flow cytometry using the fluoroprobe dihydrorhodamine 123 (DHR) in LPS-primed macrophages.
Effect of APN on LPS-induced autophagy
LPS (500 ng/mL) was applied for 24 h to establish the sepsis model. APN (30 μg/mL) was applied for 30 min before the LPS treatment to establish the LA group, and APN (30 μg/mL) alone was used for 24 h and 30 min to establish the APN group. The autophagy inhibitor 3-MA (5 mM) was applied for 1 h 30 min before exposure to LPS to establish the L3 group and was applied for 1 h before the exposure to APN followed by LPS to establish the LA3 group.
APN regulates macrophage autophagy through the AMPK pathway
LPS (500 ng/mL) was applied for 24 h to establish the septic model, APN (30 μg/mL) was applied for 30 min before LPS treatment to establish the LA group, and APN (30 μg/mL) alone was used for 24 h and 30 min to establish the APN group. The AMPK inhibitor Compound C (10 μmol/L) was applied for 1 h 30 min before exposure to LPS to establish the leukocytes (LC) group and was applied for 1 h before the exposure to APN followed by LPS to establish the LAC group.
Western blotting
At the end of each treatment, the cells were washed twice with ice-cold phosphate-buffered saline (PBS) and were then sonicated on ice in lysis buffer (50 mM Tris-HCl, pH 8.0, containing 150 mM NaCl, 1% Nonidet P-40, 5 mM ethylenediaminetetraacetic acid (EDTA), and protease inhibitor tablets from Roche or 50 mM sodium fluoride and 25 mM glycerophosphate). The cell lysates were centrifuged at 12,000g for 10 min at 4°C, and the supernatants were collected for western blotting. The protein concentration was measured using a Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA). After being boiled for 5 min in the loading buffer with 10% 2-mercaptoethanol, samples, containing 30 μg of protein, were separated on 10% or 12% Tris-glycine gels. The separated proteins were transferred onto a nitrocellulose membrane (Bio-Rad). Immunoblotting was performed using individual antibodies.
Measurement of ROS
2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA), a ROS-specific fluorescent probe, was used to determine the intracellular ROS levels. RAW 264.7 cells were exposed to 20 μM H2DCF-DA for 30 min and were then washed twice with PBS. The level of fluorescence was assessed using a fluorescence plate reader (excitation wavelength: 485 nm; emission wavelength: 538 nm).
Statistical analysis
The data are expressed as mean ± standard error. Comparisons among multiple groups were determined by a one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test where appropriate. Values of P < 0.05 were considered statistically significant.
Results
APN suppresses the activation of NLRP3 induced by LPS
First, we determined whether APN inhibits the LPS-induced inflammatory response. RAW 264.7 cells were incubated with LPS (500 ng/mL) for 24 h to establish a model of sepsis. APN (30 μg/mL) was applied for 30 min before LPS treatment. After the cells were exposed to LPS, the levels of NLRP3 and IL-1β were significantly increased. However, APN reversed this effect (Figure 1(a)). We then tested whether the level of NLRP3 changes after exposing the cells to an NLRP3 inhibitor. In an in vitro experiment, the NLRP3 inhibitor glibenclamide (50 μM) was preincubated with macrophages for 30 min before the LPS treatment. The levels of NLRP3 and IL-1β, after exposure to LPS, were significantly increased. However, a pretreatment with glibenclamide markedly attenuated the changes in IL-1β, suggesting that NLRP3 activation mediated by LPS induced the release of IL-1β, an effect that would be weakened by an NLRP3 inhibitor. The preincubation of macrophages with APN 30 min before the LPS treatment decreased the levels of NLRP3 and IL-1β (Figure 1(b)).
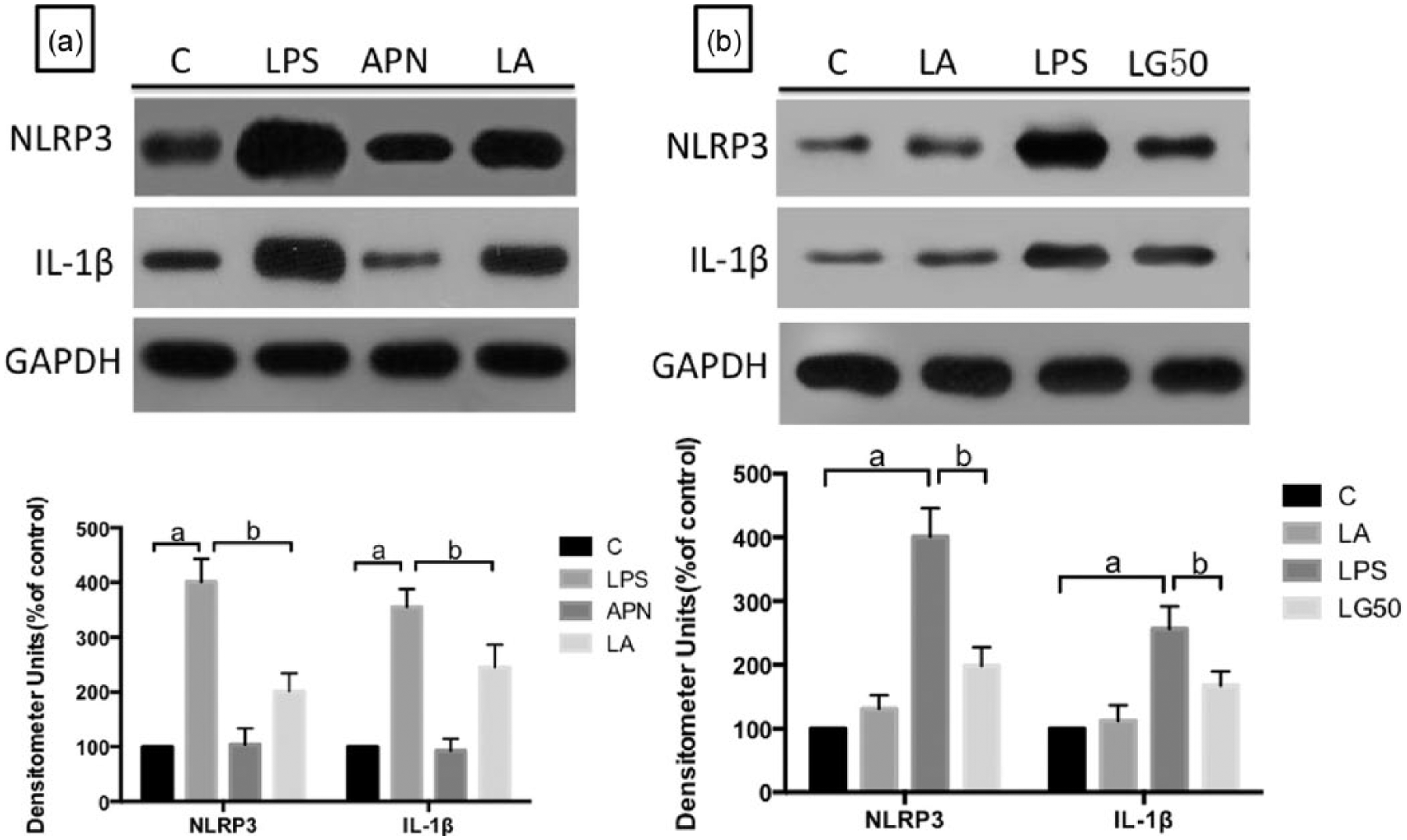
Adiponectin suppresses the activation of NLRP3 induced by LPS: a) cells cultured in regular medium were incubated with LPS (500 ng/mL) for 24 h to establish the LPS group. In addition, adiponectin (30 μg/mL) alone was used for 30 min to establish the APN group. Additionally, adiponectin (30 μg/mL) was applied for 30 min before LPS treatment to establish the LA group. (b) The cells were cultured in regular medium were incubated with LPS (500 ng/mL) 24 h to set up LPS group. In addition, adiponectin (30 μg/mL) alone was applied for 30 min to establish the APN group. Glibenclamide (50 μM) was applied for 30 min before the LPS treatment. These experiments were repeated three times, and three wells were used for each treatment. The levels of NLRP3 and IL-1β were confirmed by immunoblotting. The data are presented as mean ± SD.
APN regulates NLRP3 activation by modulating ROS production
ROS is essential for inflammasome activation. 13 The treatment of macrophages with LPS increased ROS production and the level of NLRP3, and this response was inhibited by APDC, an ROS inhibitor. However, it was aggravated when the Complex I inhibitor rotenone was applied. Since excessive ROS production is a critical event for development of inflammation, we used flow cytometry to investigate whether APN inhibits LPS-induced ROS production in macrophages. LPS treatment enhanced ROS production in a manner that was consistent with previous results, but the pretreatment with APN significantly suppressed LPS-induced ROS production in RAW 264.7 macrophages and decreased the level of NLRP3. However, the Complex I inhibitor rotenone weakened this effect (Figure 2(a) and (b)).

Adiponectin regulates NLRP3 activation by modulating ROS production: (a) ROS production was determined by flow cytometry using the fluoroprobe dihydrorhodamine 123 (DHR) in LPS-primed macrophages pretreated with the ROS inhibitor APDC (50 μM) and the Complex I inhibitor rotenone (5 μM). Additionally, adiponectin (30 μg/mL) was applied 30 min before the LPS treatment. MFI stands for the “mean fluorescence intensity.” (b) LPS (500 ng/mL) was applied for 24 h to establish the LPS group, adiponectin (30 μg/mL) was applied for 30 min before the LPS treatment to establish the LA group, the ROS inhibitor APDC (50 μM) was applied for 30 min before treatment with adiponectin followed by LPS to establish the LAA group, and the Complex I inhibitor rotenone (5 μM) was applied for 30 min before treatment with adiponectin followed by LPS to establish the LAR group. The levels of NLRP3 were confirmed by immunoblotting. The data are presented as mean ± SD.
Effect of APN on LPS-induced autophagy
Because macrophage autophagy is thought to be the main method by which ROS are eliminated, we next investigated whether APN affects autophagy to reduce the level of ROS. Immunohistochemistry with anti-LC3 showed that the expression of LC3 protein, an autophagosome marker, was higher in the APN and LPS groups than in the C group. However, the expression in the LPS group was lower than that in the APN group. At the same time, the level of ROS was negatively correlated with autophagy in the different groups. Interestingly, when the macrophages were pretreated with the autophagy inhibitor 3-MA, the function of APN in suppressing LPS-induced ROS production was inhibited (Figure 3(a) and (b)).

Effect of adiponectin on LPS-induced autophagy: (a) LPS (500 ng/mL) was applied for 24 h to establish the sepsis model. Adiponectin (30 μg/mL) was applied for 30 min before LPS treatment to establish the LA group, and adiponectin (30 μg/mL) alone was used for 24 h and 30 min to establish the APN group. The autophagy inhibitor 3-MA (5 mM) was applied for 1 h and 30 min before exposure to LPS to establish the L3 group and was applied for 1 h before the exposure to adiponectin followed by LPS to establish the LA3 group. These experiments were repeated three times, and three wells were used for each treatment. The levels of LC3I and LC3II were confirmed by immunoblotting. (b) ROS production was determined by flow cytometry using the fluoroprobe dihydrorhodamine 123 (DHR) in each group. The data are presented as mean ± SD.
APN regulates macrophage autophagy through the AMPK pathway
AMPK is downstream of APN and plays a critical role in autophagy induction. 14 To determine whether APN modulates autophagy through the AMPK pathway, we conducted in vitro experiments and examined the effect of APN and Compound C, an inhibitor of AMPK, on AMPK and LC3 expression (Figure 4(a) and (b)). LPS and APN increased the expression of AMPK and LC3, and a pretreatment with Compound C reversed these effects (Figure 4). These results suggest that APN might induce autophagy in macrophages through the AMPK pathway.

APN regulates macrophage autophagy through the AMPK pathway: (a) LPS (500 ng/mL) was applied for 24 h to establish the septic model. Adiponectin (30 μg/mL) was applied for 30 min before the LPS treatment to establish the LA group, and adiponectin (30 μg/mL) alone was used for 24 h and 30 min to establish the APN group. The AMPK inhibitor Compound C (10 μmol/L) was applied for 1 h and 30 min before exposure to LPS to establish the LC group and was applied for 1 h before the exposure to adiponectin followed by LPS to establish the LAC group. These experiments were repeated three times, and three wells were used for each treatment. The level of AMPK was confirmed by immunoblotting. (b) The levels of LC3I and LC3II were measured in each group by immunoblotting. The data are presented as mean ± SD.
Discussion
Sepsis is a major healthcare problem that is associated with long hospitalization times and poor outcome. 15 Recent studies show that ROS is involved in the process of sepsis.16,17 ROS may activate the NF–κB (nuclear factor kappa-light-chain-enhancer of activated B cells) signaling pathway, after which NF–κB may induce NLRP3 expression 18 and increase the release of inflammatory cytokines. 19 LPS treatment causes a significant increase in ROS production, which is a critical event for the development of many pathophysiologic conditions. Macrophages are the main cellular source responsible for LPS-induced ROS production, and excessive ROS production by macrophages leads to oxidative stress during the process of inflammation. In this study, when we applied LPS (500 ng/mL) for 24 h to establish a sepsis model, we detected a relationship between mitochondrial ROS activity and the presence of NLRP3 in the supernatant of the RAW 264.7 macrophages.
To avoid cellular damage, cells constantly remove ROS-generating damaged mitochondria through autophagy.19,20 In agreement with this established mechanism of mitochondrial disposal, the level of the autophagy-associated protein LC3 increased after the addition of LPS. Therefore, we speculated that inhibition of autophagy would increase the number of ROS-producing damaged mitochondria, which would activate the NLRP3 inflammasome. In this study, LPS was added to RAW 264.7 macrophages, and this resulted in increased concentrations of LC3 and of ROS. These effects were blocked by the antioxidant APDC. However, they were aggravated when the Complex I inhibitor rotenone was applied, and the level of NLRP3 showed a negative correlation with ROS. It is therefore feasible that a functional autophagy system might act as a scavenger of mitochondrial ROS through the removal of damaged mitochondria and might thereby suppress NLRP3 inflammasome activation in sepsis.
APN, the most abundant adipokine in plasma, is implicated in a number of beneficial processes. It is a major adipokine with anti-inflammatory properties, and it inhibits the proliferation of cancer cells, 21 plays a potential protective role against the ill effects of chronic alcohol consumption, 22 and prevents ROS production in response to various stimuli. Although it is widely accepted that ROS production in macrophages is a critical step leading to the various pathologic conditions caused by LPS exposure, the effects of APN on LPS-induced ROS production in macrophages are not clearly understood. In this study, we examined the nature and potential mechanisms of those effects. Herein, we provided evidence that APN inhibited ROS production in an LPS-induced model of sepsis and furthermore suppressed the expression of inflammatory factors. We next investigated the mechanism by which APN decreased the level of ROS. Additional mechanistic studies demonstrate that ROS-generating mitochondria are constantly removed by autophagy.19,20 Consequently, we decided to test whether APN regulates autophagy. The results suggested that APN improved the level of LC3, an autophagy marker, and subsequently lowered the level of ROS.
AMPK is a serine/threonine protein kinase that acts as a master sensor of cellular energy balance in mammalian cells by regulating glucose and lipid metabolism and is widely thought to regulate autophagy. 23 AMPK is downstream of APN and plays a critical role in autophagy induction. To determine whether APN modulates autophagy through the AMPK pathway, we employed Compound C, an inhibitor of AMPK, to determine whether AMPK is involved in the regulation of autophagy. Interestingly, we found that LPS and APN both increased the level of AMPK, and the level was higher in the APN group, showing interesting consistencies with the level of autophagy. However, there was a significant difference in the level of ROS. We assume that the LPS-induced increase in AMPK level, which resulted in autophagy, was insufficient to remove ROS and therefore led to the inflammatory response. However, APN significantly increased AMPK levels together with autophagy and subsequently regulated the inflammatory response and APN-stimulated AMPK activation. The mechanism remains to be determined, perhaps using APN knockout mice.
In summary, we uncovered the autophagy-promoting role of APN in RAW 264.7 macrophages, and we showed that it alleviates the inflammatory reaction and reduces the levels of ROS and NLRP3 in an LPS-induced model of sepsis. The activation of autophagy is a key mechanism by which APN might contribute to treating sepsis.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by a grant from the National Natural Science Foundation of China (81401620) and a grant to HGB from the Jiangsu Provincial Special Program of Medical Science (BL2014012). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.