
Abstract
Diclofenac sodium is widely used for its anti-inflammatory and analgesic effects in the symptomatic treatment of acute and chronic inflammatory and painful conditions. The present observational study was carried out to investigate efficacy, safety, and tolerability of a new subcutaneous (SC) 75 mg/1 mL diclofenac hydroxypropyl-β-cyclodextrin (HPβCD) formulation for the treatment of neuropathic pain both in patients without cancer and in patients with cancer. A total of 105 outpatients and inpatients with moderate to severe neuropathic pain related to cancer (CP) and non-cancer (NCP), were selected if Numeric Rating Scale (NRS) ⩾7 and Pain Detect (PD) >16, and treated with 75 mg/1 mL SC HPβCD, according to a real clinical setting. The analgesic efficacy of the treatment, which was assessed during the study by questionnaire, was expressed in terms of reduction of 30–50% and >50% of baseline pain (NRS), after 1.5 h and 4 h from drug administration; tolerability and safety were assessed as well, and adverse events were recorded by an expert panel. In our study, a significant reduction in pain intensity (PI) after 1.5 h and after 4 h was observed for both groups of patients (CP and NCP). Different from what was detected in the CP group, a further and significant reduction of PI was obtained in the NCP group at 4 h (P <0.039). With regard to tolerability, while 90% of the patients did not complain of any adverse drug reactions, only 10% reported mild transient adverse reactions. SC administration of HPβCD has been proven effective and safe in the treatment of patients with moderate to severe neuropathic pain related to cancer or not, in a real clinical setting.
Introduction
Diclofenac is a benzene-acetic acid derivative that relieves pain and reduces inflammation.1 –5 Its anti-inflammatory activity, and most of its other phar-macological effects, are due to inhibition of prostaglandin synthesis. 6 Diclofenac strongly inhibits cyclo-oxygenase isoforms in vitro and in vivo, thus decreasing the synthesis of prostaglandins, prostacyclin, and thromboxane products. Indeed, in animals and humans its administration is followed by reduced concentrations of various prostaglandins in urine, gastric mucosa, and synovial fluid. 6
In terms of analgesic effects, its advantages are represented by a fast onset and a long duration of action.
Diclofenac is widely considered as one of the few first line NSAIDs (non-steroidal anti-inflammatory drugs) in the treatment of acute and chronic painful and inflammatory conditions such as rheumatoid arthritis, osteoarthritis, and low back pain. Furthermore, it can be used to treat acute pain and inflammation associated with minor surgery, including orthopedic and dental interventions. 7
The therapeutic index of diclofenac is generally safe in animals, being variable versus other NSAIDs according to the model used. 8 However, controlled studies in healthy subjects showed that standard dosages of diclofenac are associated with less gastrointestinal damage than aspirin. 9
It can be administered in several forms other than the oral one (topical, rectal, intramuscular, i.v.), which is particularly useful when treating patients who are unable to take oral formulation. Recently IBSA Institut Biochimique SA has developed a new 75 mg diclofenac sodium formulation (Inforce® [other trademarks: Akis®, Dicloin®) with the addition of hydroxypropyl-β-cyclodextrin (HPβCD) as complexing agent to reduce the injection volume to 1 mL, which allows a new way of administration, the subcutaneous (SC). SC formulation provides various advantages such as simple method of execution, possibility of self-administration, poor invasivity, and it represents a good alternative in those cases when oral administration cannot be used.
In this context we set out to investigate the efficacy and tolerability of SC 75 mg/1 mL diclofenac HPβCD formulation in a real clinical setting of patients suffering from acute and chronic painful/inflammatory conditions.
Methods
Study design
The study was designed as a monocenter, prospective, observational study, to be carried out at the Pain Center of Cosenza (Italy).
Patient population
Main inclusion criteria were: age >18 years; presence of acute or chronic neuropathic pain (defined by Pain DETECTE >16); moderate to severe pain defined by NRS ⩾7; and written informed consent. A total of 105 outpatients and inpatients patients who met all eligibility criteria were enrolled in the study.
In order to determine the prevalence of neuropathic pain screening Pain DETECTE questionnaires were used. 11
In Pain DETECTE questionnaires the patient easily fills in nine items that do not require clinical examination.
There are seven items that accurately describe the sensitivity and two items relating to the characteristics of radiating pain and the time course of the single type of pain.
Exclusion criteria were: (1) gastrointestinal, coagulation, hepatic, renal, cerebrovascular, cardiac, arterial disorders; (2) clinically significant or unstable concurrent diseases that could be negatively affected by NSAID administration; (3) history of alcohol or drug abuse within the previous 12 months; (4) pregnancy or breast-feeding; (5) clinically significant laboratory abnormalities detected within 30 days before inclusion; (6) any concomitant analgesic or other concomitant medication that may interact with diclofenac or affect safety; (7) pregnancy and child-bearing; (8) history of hypersensitivity to diclofenac or other NSAIDs or to one of the study medication components; (9) participation at any other clinical study in the previous 3 months; and (10) psychiatric disorders or any other clinical condition judged not adequate by the investigator.
Women of child-bearing potential were required to have a negative urine pregnancy test and to use appropriate contraception and negative nursing throughout the study.
All patients were required to attend a screening visit and undergo laboratory tests within 30 days before inclusion in the study.
With respect to their baseline painful condition, the patients were classified as follows: 53 patients (50%) with pain from radiculopathy; 28 patients (27%) with pain related to cancer; nine patients (9%) with herpes zoster neuropathy (NPH); nine patients (9%) with post-traumatic and postsurgical pain; four patients (4%) with trigeminal neuralgia (TN); one patient (1%) with diabetic neuropathy; and one patient (1%) with central neuropathic pain.
Patients were divided into two groups: non-cancer pain (NCP); and cancer pain (CP).
Medication
Patients developing Pain DETECTE >16 and NRS ⩾7 received one or two daily SC injections of 75 mg/1 mL HPβCD either in the upper part of the thigh, abdomen, or upper arm. Patients remained at the investigational site for a total of 4 h after the injection.
Evaluation of pain intensity (PI) was given according to the NRS at 1.5 h, and at 4 h after administration through a questionnaire issued to outpatients or asking inpatients directly; NRS evaluation was made at each scheduled control visit, for the whole duration of the study.
Patients belonging to the NCP group received one or two daily SC injections of 75 mg/1 mL for 2 weeks, while patients belonging to the CP group received the same treatment for 2 months.
In order to assess the tolerability profile during the follow-up period, control visits were differently scheduled, in addition to baseline visit (V1), for both groups.
NCP patients group: V2 (day 2, after 18 to 24 h from baseline), V3 (after 1 week), and V4 (after 2 weeks).
CP patients group: V2 (day 2 after 18 to 24 h from baseline), V3–V10 each week until 2 months after baseline.
In case of insufficient pain relief after two doses of the study drug, the patient was withdrawn and received an alternative therapy.
Patients were provided with patient diary and they were asked to record their PI.
Endpoints
The primary endpoint of this study was the reduction of PI expressed in terms of reduction of 30–50% and >50% of baseline pain at 1.5 h and at 4 h, after treatment with subcutaneous (SC) 75 mg/1 mL of HPβCD, measured by NRS, in both the NCP and CP groups. The primary endpoint was evaluated at each time point of the study.
Safety endpoints were measured by assessment of adverse events at any time during the study, vital signs (arterial blood pressure, heart rate), and laboratory parameters (at screening visit and final visit).
Sample size
A reduction in NRS of approximately 30% from baseline pain is generally accepted as a meaningful reduction in acute and chronic pain trials. A sample size of 120 patients was considered adequate to observe at least a 30% reduction of PI versus baseline in both NCP and CP groups. Drop-out ratio was set at 15%.
Statistical methods
Patients were stratified into two main groups: non-cancer pain (NCP) and cancer pain (CP), and the results obtained were compared.
For the absolute change in pain from baseline within groups, the comparison was done using the Wilcoxon test for paired data.
Comparisons between the two groups were made using the Mann-Whitney test for continuous variables, the X2 or Fisher’s exact test for categorical variables.
As far as the variable of efficacy (percentage reduction of pain) is concerned, comparison between the two groups (CP and NCP) was carried out using a model of analysis of variance (ANOVA), ‘raw’ and ‘adjusted’ being inserted as covariates, in addition to the group, the pain at baseline, and the time to persistence of pain.
The last observation carried forward technique was used to deal with missing data and early study discontinuations.
All efficacy endpoints were analyzed in the intention-to-treat (ITT) population (all enrolled patients who received study medication with at least one post-baseline efficacy evaluation within 1.5 and 4 h post dose). The primary endpoint was also evaluated in the per-protocol (PP) population (all patients in the ITT population who also fulfilled all the eligibility criteria and did not show any major protocol violations, and with evaluation of primary endpoint at each time point according to study protocol).
Safety analyses were conducted in the safety population, defined as all enrolled patients who received at least one dose of study medication.
Statistical analyses were performed using SAS (SAS Institute Inc., Cary, NC, USA). Software, release 9.2 TS Level 02m0, on a Window XP Pro operating system.
For all tests the significance level was set at 0.05 (α).
Results
One hundred and five patients were enrolled from October 2013 to February 2014 at the Pain Center of Cosenza and the typology of neuropathic pain was reported as follows: pain from radiculopathy (n = 53); pain related to cancer (n = 28); herpes zoster neuropathy (n = 9); post-traumatic and postsurgical pain (n = 9); trigeminal neuralgia (n = 4); diabetic neuropathy (n = 1); and central neuropathic pain (n = 1) (Figure 1).
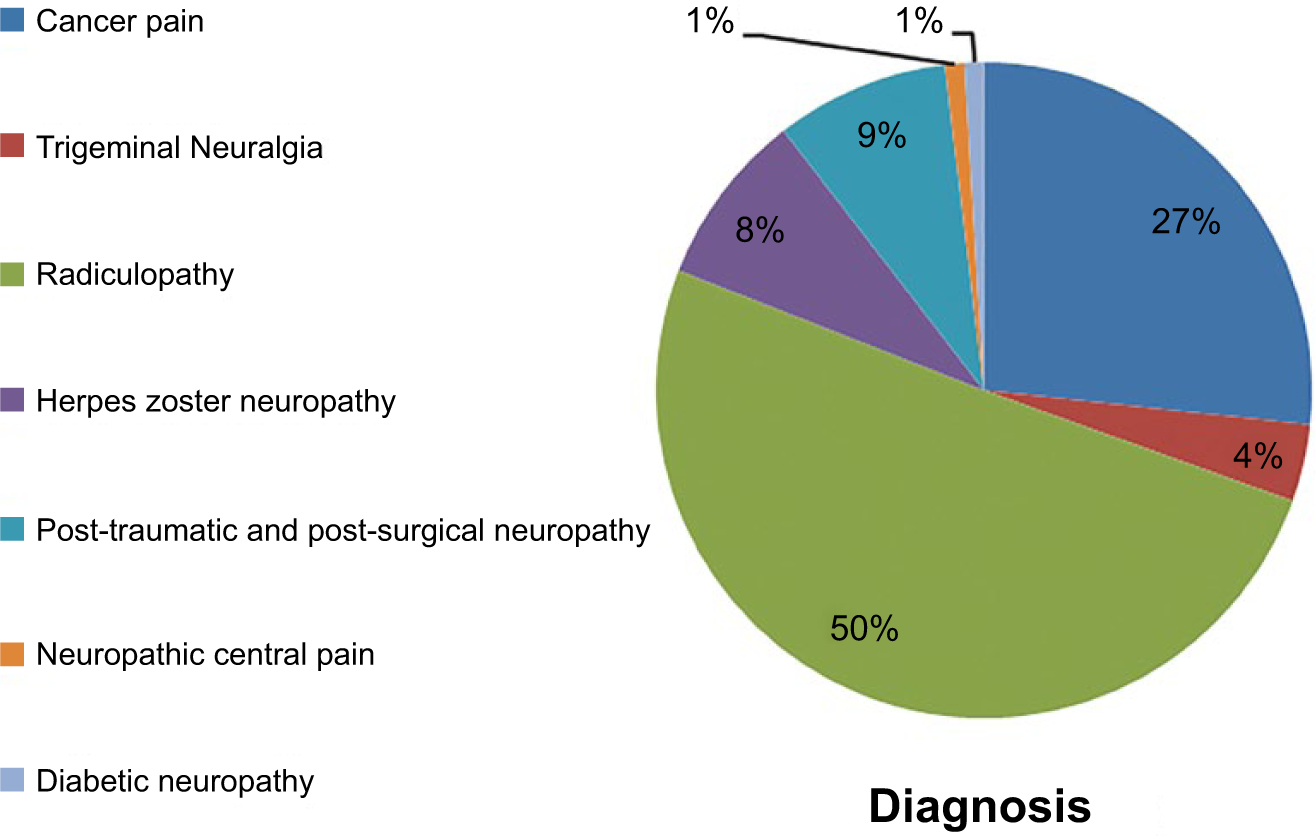
Typology and percentage of pain diagnosed at baseline.
All patients enrolled were stratified into two main groups: NCP (n = 77, 73%) and CP (n = 28, 27%).
All 105 enrolled patients (women, 59%), which received study medication and had at least one post-baseline evaluation, were included in ITT population. A total of 15 patients were considered as major protocol violators; thus PP population was constituted by a sample of 90 patients.
By comparing the information collected in relation to the two groups of ‘diagnosis’, NCP and CP, there were no significant differences in the distribution by age, time of persistence of pain, and pain detected.
Mean age at baseline was 59.4 years and 65.4 years in the NCP and CP groups, respectively (Table 1), while mean pain detected at baseline was 27. 8 and 27.1 in the NCP and CP groups, respectively (Table 2).
Age.Mann-Whitney test for comparison between the two groups (NCP vs. CP); P = 0.09.

Pain detected.Mann-Whitney test for comparison between the two groups (NCP vs. CP); P = 0.581.

The mean PI at baseline was significantly higher (8.55 vs, 8.11, P = 0.039) in the NCP group vs. CP group, probably due to the minor use of concomitant analgesic therapy by patients not suffering from cancer pain (Table 3).
Pain intensity at baseline.Mann-Whitney test for comparison between the two groups (NCP vs. CP); P = 0.039.

At baseline, duration of pain was in most of the patients (n = 58, 55%) for at least six months (Figure 2).

Time of onset of pain.
Efficacy endpoints
In the ITT population, the results obtained in the present study showed a significant reduction of PI in both NCP and CP patients, at 1.5 h and at 4 h.
Notably, the reduction of PI in CP group at 4 h remained almost stable, while in the NCP group a further reduction of PI was observed (Figure 3); in Table 4, confirmation of better efficacy of the study drug in patients without cancer was provided by the percentage reduction in the intensity of raw PI, and after adjustment at different times, which was significantly higher in NCP group at 4 h (P = 0.039) vs. CP group (Table 4).

Significant reduction in PI at 1.5 h and at 4 h. While the reduction of PI in CP group at 4 h remains almost stable, in the NCP group it shows a further reduction.
Percentage reduction in the raw PI and adjusted at different times (Anova).

It should be noted that the mean values of PI reduction obtained after administration of SC 75 mg diclofenac at day 1 (see Figures 3 and 4) kept a similar profile at any time point, in both the subgroups examined, through the whole duration of the study.

The percentage of PI reduction for the group at 1.5 h and at 4 h after administration of study drug.
Data of PP population (not shown) were comparable to data obtained in ITT population.
Safety and tolerability
No serious adverse event occurred during the study. Ninety-four patients (90% of the study population) did not complain of any adverse drug reactions, while only 11 patients (10% of the study population) reported mild transient adverse drug reactions, mostly dyspepsia and skin reactions, which resolved spontaneously (see Table 5 and Figure 5).
Adverse events developed after subcutaneous administration of diclofenac 75 mg/1 mL.


Adverse events developed after subcutaneous administration of diclofenac 75 mg/1 mL.
Local tolerability of the SC injections was considered excellent or good in more than 90% of the patients. Neither clinically significant change in laboratory parameters nor in vital signs was observed from screening to the final visit in both groups of patients.
Discussion
The current study showed that SC diclofenac HPβCD at 75 mg produced a higher pain intensity decrease at 1.5 h and at 4 h post dose, versus pain intensity registered at baseline, both in NCP and in CP patients. Notably, a further and significant percentage reduction of pain was detected at 4 h post dose in NCP patients, but not in CP patients whose decrease of pain was similar to that observed at 1.5 h.
With respect to the reason of the higher efficacy of SC 75 mg in NCP patients vs. CP patients at 4 h post dose, it should be highlighted that more pathogenetic mechanisms may play a central role and cause pain in patients with cancer. Second, patients suffering from cancer represented 25% of the patient population, all of them taking opioids as standard pain therapy.
Therefore, the results obtained in this study confirm the analgesic effect of SC administration of diclofenac 75 mg as already previously demonstrated in patients suffering from moderate to severe acute pain.4,7
Regarding safety, the data of the present investigation are comparable with the safe profile seen in previous studies, with 90% of patients population free of any adverse drug reaction and only 10% of patients showing mild transient adverse reactions. The excellent profile of tolerability of SC 75 mg diclofenac, which was kept up to 2 weeks and 2 months in NCP and CP patients, respectively, can guarantee an high profile of therapy adherence and consequently a satisfactory pain control. Moreover, it should also be stated that no serious adverse event has been reported during the whole duration of the study.
As far as the formulation of diclofenac used in the current study is concerned, it should be remarked that, although no direct comparison vs. other NSAIDs is presently available, in a previous published study the single dose of SC diclofenac 75 mg/1 mL showed to be equivalent to a single intramuscular injection (IM) dose of the marketed product Voltarol 75 mg/3 mL, in healthy subjects. 12
Moreover, it should be remarked that subcutaneous administration of diclofenac 75 mg /1 mL represents an easier and potentially safer procedure versus IM administration, being less likely both blood vessel piercing and nerve damage.
Last but not least, differently from IM administration, the SC formulation used in the present study can be taken into account as self-administration as well, if required.
For the above mentioned reasons, the result of this study provide support for the analgesic efficacy of subcutaneous injection of 75 mg/1 mL diclofenac HPβCD both in NCP patients and in CP patients.
Finally, the data obtained in this study suggest that the new subcutaneous administration of Diclofenac at a dose of 75 mg can be considered a valid aid to therapy also in patients treated with long-term opioids.
Patients in the CP subgroup were 25% of NCP subgroup and this could be a limitation when analyzing the intergroup results of the study.
Administration of study drug twice daily, which was required for some patients, might be a further limitation, although in both groups of patients efficacy data obtained after the first administration of SC diclofenac 75 mg kept stable during the whole follow-up of the study.
However, due to the observational design of the study, it should be remarked that the above mentioned limitations should not be considered as critical issues.
The results of this study demonstrate that the new SC formulation of diclofenac 75 mg/1 mL produces significantly pain relief both in NCP patients and in CP patients after 1.5 h and after 4 h.
Reduction of pain in the CP group at 4 h remains almost stable, while a further and significant reduction of pain is detected in the NCP group.
No adverse events have been reported in 90% of the patient population, while in 10% adverse events are mild and transient
SC administration of diclofenac 75 mg is effective and well tolerated for relieving neuropathic pain both in patients with pain not associated to cancer and in patients with pain associated to cancer.
In conclusion, data from the current study suggest that SC administration of the new diclofenac 75 mg/1 mL formulation can represent an additional tool for the treatment of patients affected with acute and chronic neuropathic pain. 13
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.