
Abstract
Objective
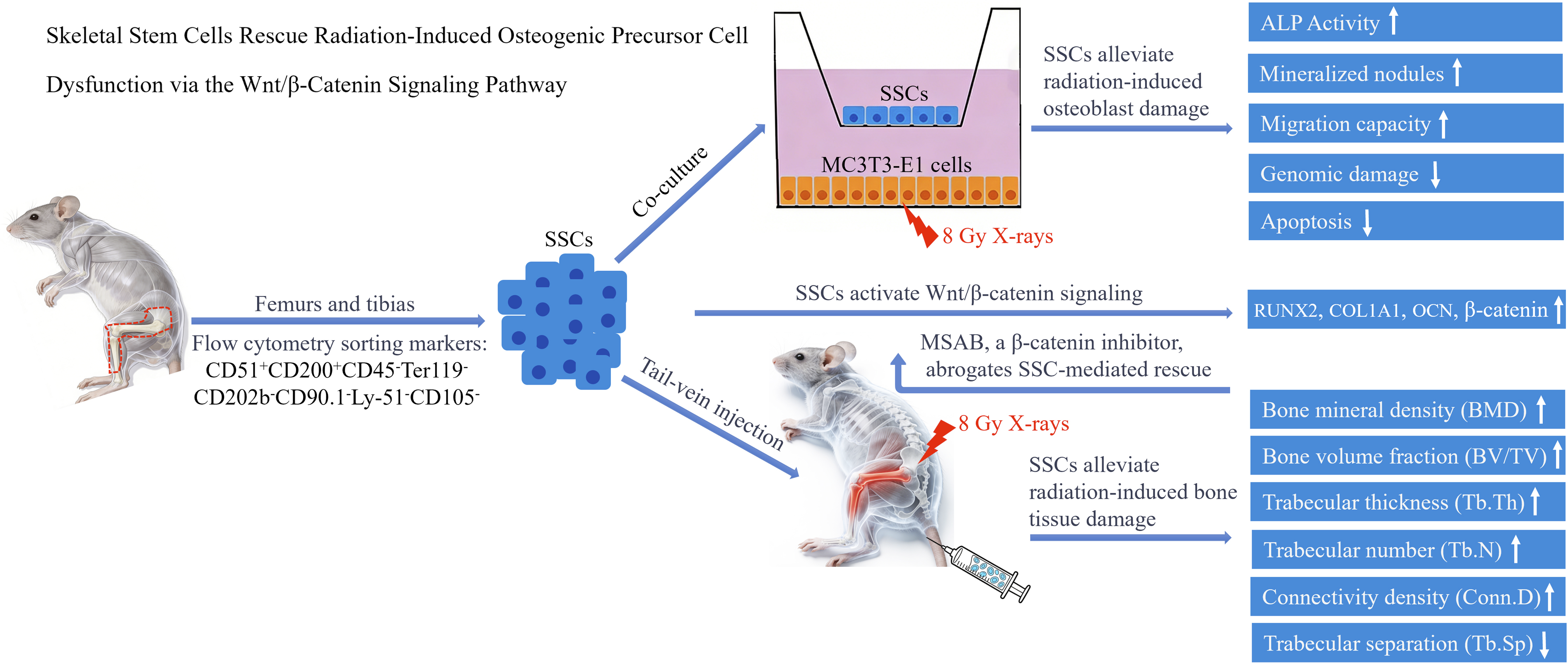
Skeletal stem cells (SSCs), a specialized subset of mesenchymal stem cells, drive bone regeneration. This study aimed to characterize the pathological effects of ionizing radiation (IR) on osteogenic precursor cells (MC3T3-E1) and to elucidate the therapeutic efficacy and underlying molecular mechanisms of SSC-mediated rescue.
Methods
Experiments were performed using clonogenic assay, micronucleus assay, flow cytometry, osteogenic differentiation assessment, migration assay, and animal model establishment.
Results
The results demonstrated that IR compromised the clonogenic capacity of MC3T3-E1 cells, exacerbated genomic damage, induced apoptosis, and significantly impaired their osteogenic differentiation and migratory potential. In vivo, IR led to deterioration of the skeletal microarchitecture,as evidenced by reduced bone mineral density (BMD), decreased bone volume fraction (BV/TV), and a concomitant increasee in marrow adiposity and osteoclastogenesis. Notably, SSC treatment significantly reversed these deleterious effects in both cellular and animal models. Mechanistically, SSC intervention upregulated key osteogenic regulators (RUNX22, COL1A1, and OCN) and stabilized β-catenin. Crucially, the radioprotective efficacy of SSCs was abrogated by the Wnt/β-catenin inhibitor MSAB, demonstrating the pathway’s indispensable role in the rescue process.
Conclusions
This study indicates that SSCs effectively alleviate IR-induced injury to osteoblast progenitor cells, providing compelling evidence and establishing a novel therapeutic paradigm for treating radiation-related skeletal complications.
Keywords
Introduction
Radiotherapy is a highly effective treatment modality for malignant tumors located in the pelvic region, spine, and extremities. However, its cytotoxic effects are non-specific, often damaging adjacent healthy tissues, particularly bone. Bone tissue absorbs 30–40% more radiation than surrounding soft tissues due to its high calcium content, making it a common site of severe secondary injury in cancer survivors. 1 A significant long-term complication is the irreversible deterioration of bone quality, which can lead to fractures, osteopenia, and osteoradionecrosis. 2 The pathogenesis of this injury is multifactorial, primarily driven by direct impairment of the osteogenic lineage—most notably skeletal stem cells (SSCs) and their descendant osteogenic precursor cells (OPCs). 3 Radiation induces apoptosis, cellular senescence, and a significant loss of osteogenic differentiation potential in these cells, thereby compromising the innate regenerative capacity of bone. 4
Cell-based therapies, particularly those using mesenchymal stem cells (MSCs), have emerged as a promising strategy in bone tissue engineering and regenerative medicine. MSCs have been extensively studied for their therapeutic potential in skeletal regeneration, due to their ability to differentiate into osteoblasts, chondrocytes, and adipocytes in vitro. 5 However, MSCs are inherently heterogeneous populations isolated based on plastic adherence and expression of broad-spectrum surface markers —such as CD73, CD90, and CD105—rather than lineage-specific properties; therefore, it is difficult to isolate a pure, uniform population of functionally competent stem cells.6,7 This heterogeneity leads to inconsistent functional potency: MSC cultures consist of a mixture of stem cells, progenitors, and differentiated cells, and thus exhibit variable capacity to support bone repair. Moreover, MSCs can differentiate into non-skeletal lineages such as myocytes and fibroblasts, but their therapeutic application is limited by immune rejection, poor storage stability, and potential tumorigenic risks. 8 These limitations highlight the need for a more precisely defined, lineage-restricted stem cell population for targeted skeletal regeneration.
SSCs represent a breakthrough as they are a rare cell population superior to MSCs, defined by strict functional and phenotypic criteria. Unlike heterogeneous MSCs, SSCs can be prospectively isolated using a specific combination of surface markers, enabling purification of a homogeneous cell population.9,10 Functionally, SSCs exhibit two defining characteristics that distinguish them from MSCs. First, they are lineage-restricted, giving rise exclusively to bone, cartilage, and stromal tissues, while lacking the capacity to differentiate into adipogenic, myogenic, or fibroblastic lineages. This specificity ensures that their regenerative potential is precisely directed toward skeletal tissue repair and maintenance. Second, they exhibit robust self-renewal capacity, as evidenced by serial clonogenic formation in vitro and the ability to reconstitute multilineage ossicles containing bone, cartilage, and marrow cavities following serial transplantation in vivo.11,12 Moreover, SSCs exhibit tissue-specific regenerative potential, as evidenced by their localized proliferation in response to acute skeletal injuries—such as fractures—a dynamic and functionally significant response that is not consistently observed in heterogeneous MSC populations. 13
Previous studies indicate that MSCs exert their therapeutic effects not only through direct differentiation but also through paracrine regulation of neighboring damaged cells. These studies have demonstrated that MSCs secrete multiple bioactive factors to modulate signaling pathways in target cells, thereby promoting tissue repair.14,15 The Wnt/β-catenin signaling pathway plays a central role in both skeletal development and the maintenance of bone homeostasis in adults. As a key molecular cascade, it governs multiple stages of bone formation by directing the fate of MSCs toward the osteoblastic lineage. 16 Activation of this pathway promotes osteoblast differentiation from MSCs, enhances the proliferation of bone-forming cells, and protects these cells from apoptosis. 17 Interestingly, IR has been shown to disrupt Wnt/β-catenin signaling in hematopoietic and mesenchymal stem cells, thereby impairing tissue regeneration. Conversely, low-dose IR can promote osteoblast differentiation by activating Wnt/β-catenin signaling, suggesting a dose-dependent relationship between IR and this pathway. 18 Recent evidence has shown that SSCs regulate their own osteogenic differentiation via the Wnt/β-catenin signaling pathway, 19 but it remains unclear whether SSCs can modulate this pathway in IR-damaged OPCs to restore their function.
In this study, we hypothesized that SSCs can rescue the proliferative capacity and osteogenic differentiation potential of irradiated OPCs by activating the Wnt/β-catenin signaling pathway. We tested this hypothesis using in vitro co-culture experiments and pharmacological inhibition of the pathway, providing a theoretical basis for SSC-based therapy in IR-induced bone injury.
Materials and Methods
Study Duration
All experiments were conducted from October 2021 to August 2024.
Isolation and Identification of SSCs
Antibodies Used for SSC Identification

Cell Culture and Irradiation Treatment
The murine osteoblast progenitor cell line MC3T3-E1 was purchased from the Cell Bank of the Chinese Academy of Sciences. The cells were cultured in α-MEM supplemented with 10% FBS and 1% penicillin-streptomycin in a constant-temperature incubator at 37°C with 5% CO2. Cells were irradiated with 8 Gy X-rays using an RS2000 X-ray device (RS-2000 Pro, Rad Source, GA, USA) at a dose rate of 2 Gy/min. Sham-irradiated cells served as the negative control group.
Co-Culture Systems
SSCs were seeded in transwell inserts (0.4 μm pore size, Corning, NY, USA, Cat. No. 8600A51) at 2.5×104 cells/insert, and irradiated MC3T3-E1 cells were seeded in 6-well plates at 7.5×104 cells/well. Inserts were placed into the wells containing irradiated MC3T3-E1 cells, and cells were cultured in osteogenic induction medium. For pathway inhibition, 10 μmol/L MSAB (MedChemExpress, Monmouth Junction, NJ, USA, Cat. No. HY-120697), a Wnt/β-catenin signaling pathway inhibitor, was added to the co-culture medium.
Clonogenic Assay
To assess the radiosensitivity and long-term proliferative potential of MC3T3-E1 cells, a clonogenic assay was performed. Cells were seeded at graded densities (150, 200, 400, 1,000, and 2,000 cells/well) in 6-well plates. After a 24-hour adherence period, the plates were irradiated with X-rays at doses of 0, 2, 4, 6, and 8 Gy at room temperature. Following irradiation, the cells were cultured for 14 days to facilitate colony formation. The resulting colonies were rinsed twice with PBS, fixed in 70% ethanol, and stained with 0.1% crystal violet. Colonies comprising at least 50 cells were identified as viable colonies and quantified manually to determine the surviving fraction.
Micronucleus Assay
MC3T3-E1 cells were seeded into 6-well plates (1 × 105 cells/well), cultured for 24 hours, and then divided into control (non-irradiated) and 8 Gy X-ray irradiated groups. 2 hours post-irradiation, cells were treated with cytochalasin B (2.0 μg/mL) to inhibit cytokinesis and induce binucleated cell formation. After an additional 24 h, cells were fixed with 4% paraformaldehyde and stained with DAPI (Beyotime, Shanghai, China, Cat. No. C1005) to visualize nuclei and micronuclei. The frequency of micronucleus (MN) formation was quantified by counting the number of micronuclei per 1,000 binucleated cells across multiple randomly selected microscopic fields per group using a microscope (IX73, Olympus, Tokyo, Japan).
Apoptosis Assay
Cellular apoptosis was quantified using an Annexin V-PE/7-AAD apoptosis detection kit (Univ, Shanghai, China, Cat. No. abs50007) according to the manufacturer’s instructions. Briefly, MC3T3-E1 cells were harvested at 24- and 48-hours post-irradiation, washed with ice-cold PBS, and resuspended in binding buffer. The cells were then incubated with PE-conjugated Annexin V and 7-aminoactinomycin D (7-AAD) for 15 minutes in the dark. Apoptotic cells were analyzed by flow cytometry (FACSVerse, BD Biosciences, CA, USA). Cells positive for Annexin V were identified as the apoptotic population.
Transwell Migration Assay
MC3T3-E1 cells were irradiated with 8 Gy X-rays, then harvested and resuspended in serum-free α-MEM at 5×104 cells/mL. A total of 200 μL of the cell suspension was added to the upper chamber of a Transwell insert (8 μm pore size, Corning, Cat. No. 353097), and 600 μL of α-MEM containing 10% FBS was added to the lower chamber. After culturing for 24 hours and 48 hours, the Transwell insert was removed. Cells on the upper surface of the membrane were wiped off with a cotton swab, and cells on the lower surface were fixed with 4% paraformaldehyde for 30 minutes and stained with crystal violet for 15 minutes. Cells were counted under a microscope (6 random fields per insert), and the relative migration rate was calculated by normalizing to the control group.
ALP Staining and Activity Assay
MC3T3-E1 cells were seeded into 6-well plates (1×105 cells/well for staining) or 96-well plates (1×104 cells/well for activity detection). After irradiation and corresponding interventions, cells were cultured in osteogenic induction medium (iXCells Biotechnologies, San Diego, CA, Cat. No. MD-0006) for 7 days. For ALP staining, cells were rinsed twice with PBS, fixed with 4% paraformaldehyde for 15 minutes, and stained using an ALP staining kit (Abcam, Cambridge, UK, Cat. No. ab284936) following the manufacturer’s instructions. Staining intensity was observed and imaged under a microscope (IX73, Olympus). For ALP activity quantification, cells were lysed using RIPA lysis buffer, and protein concentrations were determined by the BCA method. ALP activity was measured using an ALP activity detection kit (Sigma-Aldrich, Burlington, MA, USA, Cat. No. AP0100), and the relative ALP activity was calculated.
Alizarin Red S Staining
MC3T3-E1 cells were seeded at a density of 1×105 cells per well in 6-well plates and cultured in osteogenic induction medium for 21 days after irradiation and intervention. The medium was discarded, and cells were washed twice with PBS. Cells were fixed with 4% paraformaldehyde for 30 minutes, stained with 0.1% Alizarin Red S (Beyotime, Cat. No. C0148S) for 30 minutes at room temperature, and rinsed with distilled water until no excess dye was left. The number and intensity of mineralized nodules were observed under a microscope (IX73, Olympus). For quantification, 10% cetylpyridinium chloride was added to each well to elute the dye, and the absorbance was measured at 562 nm using a microplate reader (Neo-2, Synergy, Biotek instrument, CA, USA). The relative mineralization level was calculated by normalizing to the control group.
qRT-PCR Analysis
MC3T3-E1 cells in each group were collected, and total RNA was extracted using Trizol reagent (Invitrogen, Waltham, MA, USA, Cat. No. 15596026CN) according to the manufacturer’s instructions. Reverse transcription was performed to synthesize complementary DNA (cDNA) using a reverse transcription kit (Takara, Shiga, Japan, Cat. No. RR037A). Quantitative real-time PCR (qRT-PCR) was performed using a fluorescent quantitative PCR kit (Thermo Fisher Scientific, Waltham, MA, USA, Cat. No. A25918) on Real-Time PCR System (ViiA7, Thermo Fisher Scientific), with GAPDH serving as the internal reference gene. The primer sequences were listed as follows: Runx2: Forward 5′- GCCGGGAATGATGAGAAC -3′, Reverse 5′- TGGGGAGGATTTGTGAAGA -3′. Col1a1: Forward 5′- CAGAGGCGAAGGCAACA -3′, Reverse 5′- GTCCAAGGGAGCCACATC -3′. Bglap (Osteocalcin, OCN): Forward 5′- GCTGTTTGTTCGGGTCTC -3′, Reverse 5′- GGGCCAAAGTCAGCATC -3′. GAPDH: Forward 5′- AGGTCGGTGTGAACGGATTTG -3′, Reverse 5′- GGGGTCGTTGATGGCAACA -3′. The relative expression level of each gene was calculated using the 2-ΔΔCt method.
Western Blot Analysis
MC3T3-E1 cells in each group were collected, and total protein was extracted using RIPA lysis buffer. The protein concentration was determined using a BCA protein assay kit (Thermo Fisher Scientific, Cat. No. 23225). Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 5% skim milk for 1 hour at room temperature, and then incubated with primary antibodies targeting RUNX2 (1:1000, Cell Signaling Technology, Danvers, MA, USA, Cat. No. 12556), COL1A1 (1:1000, Cell Signaling Technology, Cat. No. 72026), OCN (1:1000, Cell Signaling Technology, Cat. No. 59757), β-catenin (1:1000, Cell Signaling Technology, Cat. No. 8480), and GAPDH (1:1000, Cell Signaling Technology, Cat. No. 2118) at 4°C overnight. After washing with Tris-buffered saline with Tween 20 (TBST), membranes were incubated with horseradish peroxidase (HRP)-labeled secondary antibody (1:3000, Cell Signaling Technology, Cat. No. 7074) for 1 hour at room temperature. Protein bands were visualized using an enhanced chemiluminescence (ECL) detection kit (Cell Signaling Technology, Cat. No. 6883), and recorded utilizing a polychromatic fluorescence chemiluminescence imaging analysis system (FluorChem E, ProteinSimple, San Jose, CA, USA). The gray value of each band was quantified using Image J 1.8.0 software (Media Cybernetics, Silver Spring, MD, USA), and the relative protein expression level was determined by normalization to the corresponding GAPDH gray value.
Animal Model Establishment and Grouping
C57BL/6 mice (6-week-old, Cavens Experimental Animal Co., Ltd) were randomly divided into control group, IR group, and IR+SSC group (n=6 per group). Mice in the IR and IR+SSC groups were irradiated with a single dose of 8 Gy X-rays (targeting the hindlimb bone) using an image-guided irradiator (X-RAD SmART, Precision X-ray, North-Branford, CT, USA), while mice in the control group received no irradiation. Mice in the IR+SSC group were intravenously injected with 1×106 SSCs (suspended in 200 μL PBS) via the tail vein immediately after irradiation, and mice in the other groups were injected with an equal volume of PBS. Mice were sacrificed at 2 weeks and 4 weeks after irradiation, and the femurs were harvested for subsequent detection.
Micro-CT Analysis of Bone Tissue
Isolated femurs were fixed with 4% paraformaldehyde for 24 hours, and then scanned using a Micro-CT system (SkyScan 1174, Billerica, MA, USA). The region of interest (ROI) was selected as the metaphysis of the femur. Bone-related parameters were analyzed using CT-Analyser software, including bone mineral density (BMD), bone volume fraction (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), connectivity density (Conn.D), and trabecular separation (Tb.Sp).
Histological Staining of Bone Tissue
Hematoxylin and Eosin (H&E) Staining
Fixed femurs were decalcified with 10% ethylenediaminetetraacetic acid (EDTA) for 2 weeks, with solution changes every three days, followed by dehydration, paraffin embedding, and sectioning into 5-μm-thick slices. Sections were stained using a hematoxylin and eosin staining kit (Beyotime, Cat. No. C0105S), then dehydrated, cleared, and mounted. The marrow steatosis was observed under a microscope (IX73, Olympus), and quantified by counting steatotic cells in 6 randomly selected microscopic fields.
Tartrate-Resistant Acid Phosphatase (TRAP) Staining
Paraffin sections of femurs were dewaxed to water, and TRAP staining was performed using a TRAP staining kit (Servicebio, Wuhan, China, G1050-50T) according to the manufacturer’s instructions. Osteoclasts (TRAP-positive, multinucleated cells) were observed under a microscope (IX73, Olympus), and the number of osteoclasts was counted in 6 random fields.
Immunohistochemical Staining
Paraffin-embedded femur sections were deparaffinized and rehydrated to water, and antigen retrieval was performed by boiling in citrate buffer (pH 6.0) for 15 minutes. Sections were incubated with 3% hydrogen peroxide for 10 minutes to block endogenous peroxidase activity, and then blocked with 5% bovine serum albumin (BSA) for 30 minutes at room temperature. Sections were incubated with primary antibodies targeting β-catenin (1:200, Cell Signaling Technology, Cat. No. 8480) and Osterix (1:200, Cambridge, United Kingdom, Cat. No. ab209484) at 4°C overnight. After washing with PBS, sections were incubated with HRP-labeled secondary antibody (1:500, Cell Signaling Technology, Cat. No. 7074) for 1 hour at room temperature. Sections were stained with 3,3′-diaminobenzidine (DAB) for color development, counterstained with hematoxylin, dehydrated, transparentized, and mounted. Staining intensity was observed and imaged under a microscope (IX73, Olympus). Images were analyzed with Image-Pro Plus 7.0 software (Media Cybernetics, Rockville, MD, USA) to quantify the positively stained areas, and the relative expression levels of β-catenin and Osterix were subsequently determined.
Statistical Analysis
All experiments were repeated at least 3 times, and the data were expressed as mean ± standard deviation (mean ± SD). Statistical analysis was performed using GraphPad Prism 10.0 software (GraphPad Software, San Diego, CA, USA). Differences between groups were analyzed using unpaired two-tailed Student’s t-tests. A value of p < 0.05 was considered statistically significant.
Results
Irradiation-Induced Damage and Apoptosis in Osteoblast Progenitor Cells
The colony-forming efficiency of osteogenic precursor cells was determined via clonogenic assay to evaluate the dose-dependent effects of radiation on cellular viability. As shown in Figure 1A, the number of colonies decreased progressively with the increasing irradiation doses (0, 2, 4, 6, 8 Gy). The survival fraction curve exhibited a dose-dependent decline, indicating that irradiation significantly reduced the clonogenic potential of MC3T3-E1 cells (Figure 1B). To evaluate irradiation-induced genomic damage, micronucleus (MN) formation was examined. While the control group exhibited negligible MN frequency in binucleated cells, 8 Gy irradiation triggered a marked increase in MN formation (Figure 1C). Quantitative analysis confirmed a significant elevation in the number of MN per 1,000 binucleated cells in the irradiated group relative to the control (Figure 1D). Apoptosis was quantified via flow cytometry utilizing 7-AAD/Annexin V-PE staining. We observed a time-dependent induction of apoptosis at 24- and 48-hours post-IR (Figure 1E). Quantitative analysis confirmed that apoptotic rates were significantly elevated at 24 hours and underwent a further significant increase by 48 hours compared to the control group (Figure 1F). Collectively, these findings indicate that irradiation induces significant cellular damage and promotes apoptosis in MC3T3-E1 cells. IR induces damage and apoptosis in osteoblast progenitor cells. (A) Representative images of colony formation assays showing the morphology and quantity of cell colonies after treatment with different IR doses (0, 2, 4, 6, and 8 Gy). (B) Cell survival fraction curve derived from colony formation assay. (C) Representative fluorescence images of the MN assay (Scale bar: 25 μm). Arrows indicate micronuclei. (D) Quantitative analysis of MN numbers per 1000 binucleated cells in control and IR-treated groups. (E) Representative flow cytometry scatter plots of cell apoptosis detected by Annexin V-PE/7-AAD staining. Plots include three groups: Control (no IR), IR-24 h (24 h post-IR), and IR-48 h (48 h post-IR). (F) Quantitative analysis of total apoptotic rates. All data are presented as mean ± SD, with statistical significance determined by unpaired two-tailed Student’s t-test (**p < 0.01; ***p < 0.001).
Irradiation-Induced Functional Inhibition of Osteoblast Progenitor Cells
Osteogenic differentiation was evaluated through ALP staining and activity quantification. As illustrated in Figure 2A, ALP staining intensity was notably diminished in the IR group relative to the control. Quantitatively, the relative ALP activity in irradiated cells was significantly reduced, reaching approximately 60% of control levels (Figure 2B). Mineralization capacity was further assessed via Alizarin Red S staining; representative images (Figure 2C) revealed that mineralized nodules were both fewer in number and less robust in the IR group. Correspondingly, the relative mineralization level in the IR group was markedly attenuated, representing only 30% of the control group (Figure 2D). Furthermore, cell migration was analyzed using a Transwell assay; at both 24 and 48 hours, the number of migrated cells in the IR group was evidently lower than in the control (Figure 2E). Quantitative analysis (Figure 2F) corroborated that cell motility was significantly suppressed at both time points. Collectively, these data demonstrate that IR treatment severely inhibits osteogenic differentiation and impairs the migratory capacity of these cells. Irradiation inhibits the function of osteoblast progenitor cells. (A) Representative images of alkaline phosphatase (ALP) staining in the Control and IR groups (Scale bar: 50 μm). (B) Quantitative analysis of relative ALP activity. (C) Representative images of Alizarin Red S staining (Scale bar: 100 μm). (D) Quantitative analysis of the relative mineralization level in the Control and IR groups. (E) Representative images of the Transwell cell migration assay at 24 h and 48 h in the Control and IR groups (Scale bar: 50 μm). (F) Quantitative analysis of relative cell migration. All data are presented as mean ± SD, with statistical significance determined by unpaired two-tailed Student’s t-test (*p < 0.05; **p < 0.01; ***p < 0.001).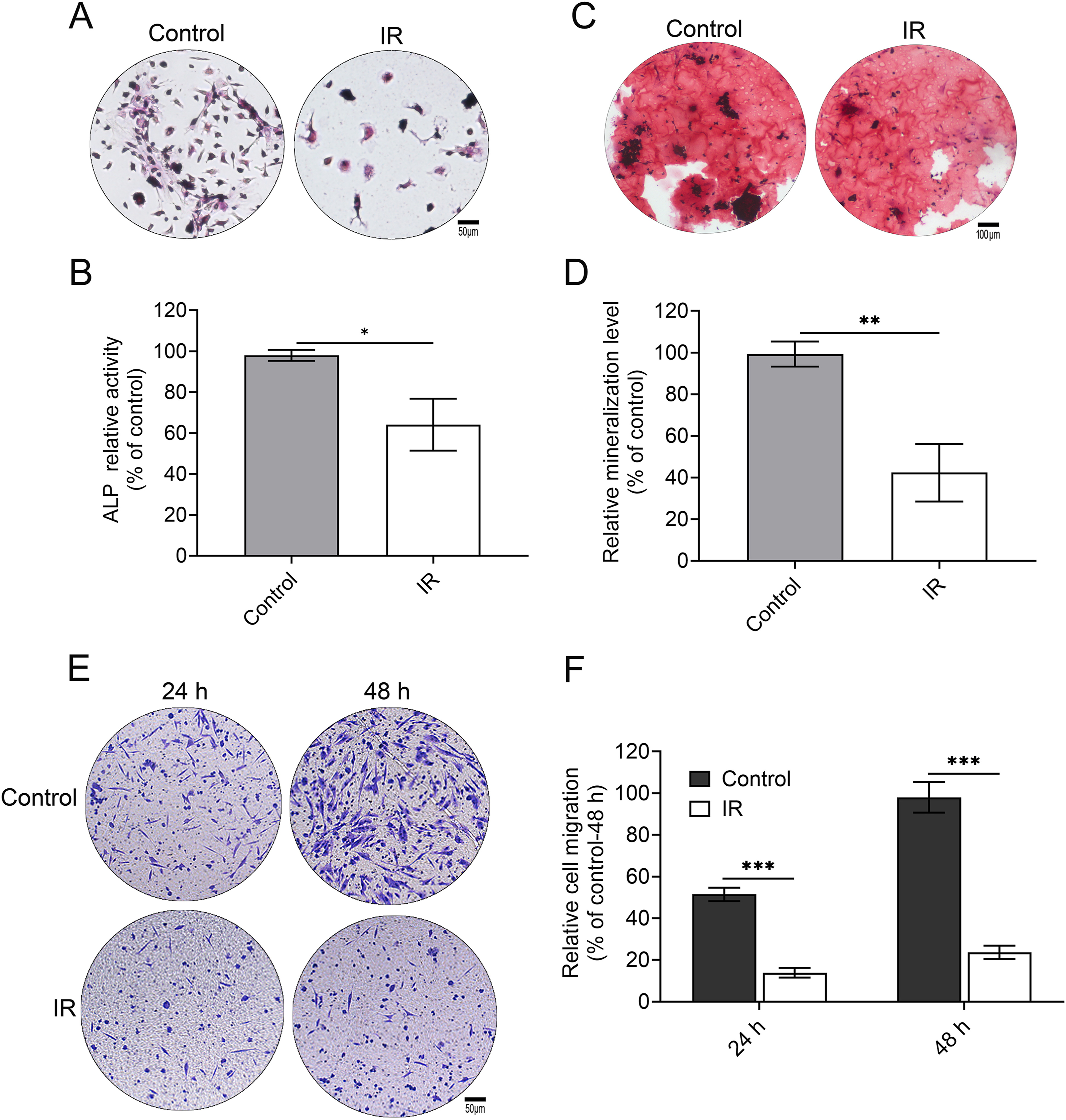
SSC Co-Culture Rescues the Function of Irradiated Osteogenic Precursor Cells
Mouse SSCs were harvested from the femurs and tibias of 6-week-old C57BL/6 mice, then phenotypically identified and purified by fluorescence-activated cell sorting (FACS) (Figure S1). To evaluate their therapeutic potential in mitigating radiation-induced injury, these purified stem cells were co-cultured with irradiated MC3T3-E1 cells. Radiation-induced apoptosis was quantified by flow cytometry. As shown in Figure 3A and 3B, the baseline apoptotic rate in the control group was low, whereas it increased markedly following irradiation. Notably, the addition of SSCs significantly alleviated this effect, with the IR+SSC group exhibiting a substantial reduction in apoptosis compared to the IR-only group. These data indicate that SSCs effectively mitigate radiation-induced programmed cell death. Representative ALP staining (Figure 3C) revealed robust activity in the control group, which was markedly suppressed following IR. Notably, SSC treatment partially rescued this effect. Quantitative assessment (Figure 3D) validated these findings, showing that SSC significantly restored ALP activity levels, thereby counteracting IR-induced inhibition. Mineralization capacity was assessed by Alizarin Red S staining. As shown in Figure 3E, the control group exhibited robust formation of mineralized nodules, which was markedly reduced in the IR group. Quantitative analysis confirmed that the relative mineralization level in irradiated cells was significantly lower than that in the control group (Figure 3F). Notably, co-culture with SSCs significantly reversed this functional deficit, as evidenced by a substantial increase in mineralization compared with the IR-only group. These findings suggest that SSCs effectively ameliorate radiation-induced impairment of mineral deposition. Transwell migration assays revealed that relative cell motility was significantly attenuated in the IR group compared to the control group. Notably, co-culture with SSCs effectively rescued this migratory deficit, as evidenced by a marked increase in the number of migrated cells relative to the IR-only group (Figure 3G and 3H). These results indicate that SSCs can effectively ameliorate radiation-induced impairment of cell migration. Co-culture with SSCs rescues the function of irradiated osteogenic precursor cells. (A, B) Cell apoptosis was analyzed by flow cytometry with Annexin V-PE/7AAD double staining. (A) Representative flow cytometry plots. (B) Quantitative analysis of the apoptotic rate. (C, D) ALP activity was assessed. (C) Representative ALP staining images (Scale bar: 50 μm). (D) Quantitative analysis of the relative ALP activity. (E, F) Mineralization capacity was evaluated using Alizarin Red S staining. (E) Representative staining images of mineralized nodules (Scale bar: 100 μm). (F) Quantitative analysis of the relative mineralization level. (G, H) Cell migration was determined by a migration assay. (G) Representative images of migrated cells (Scale bar: 50 μm). (H) Quantitative analysis of the relative cell migration level. All data are presented as mean ± SD, with statistical significance determined by unpaired two-tailed Student’s t-test (*p < 0.05; **p < 0.01).
SSCs Rescue Irradiated Osteogenic Precursor Cell Function via Wnt/β-Catenin Signaling
To determine whether the rescue effect of SSCs was pathway-specific, we treated the cells with the β-catenin inhibitor MSAB. As shown in Figure 4A and B, IR-induced suppression of ALP activity was significantly reversed by SSCs; however, this restorative effect was effectively abrogated by the addition of MSAB. This loss of function indicates that β-catenin signaling is indispensable for SSC-mediated osteogenic recovery. To evaluate mineralization potential, Alizarin Red S staining and subsequent quantification (Figure 4C and 4D) revealed that IR markedly suppressed mineral deposition. SSC intervention effectively rescued this phenotype; however, MSAB co-treatment abrogated this restorative effect. At the transcriptional level, qRT-PCR analysis (Figure 4E) showed that IR significantly downregulated the osteogenic master regulators Runx2, Col1a1, and Bglap (Osteocalcin, OCN). While SSCs robustly upregulated these markers in irradiated cells, MSAB treatment blunted this recovery. These findings were further corroborated by Western blot analysis (Figure 4F and 4G), which demonstrated that IR diminished the expression of RUNX2, COL1A1, OCN, and β-catenin. SSCs restored these protein levels, whereas MSAB effectively blocked the SSC-mediated rebound. Collectively, these data indicate that SSCs mitigate radiation-induced osteogenic dysfunction by activating the Wnt/β-catenin signaling axis, a process that is reversed by pharmacological inhibition of β-catenin. SSCs exert rescue effects via the Wnt/β-catenin signaling pathway. (A) Representative ALP staining images of cells in each group (Scale bar: 50 μm). (B) Quantitative analysis of ALP activity in each group. (C) Representative Alizarin Red S staining images of cells in each group (Scale bar: 100 μm). (D) Quantitative analysis of Alizarin Red S staining in each group. (E) Relative mRNA expression levels of osteogenic marker genes (Runx2, Col1a1, and OCN) detected by qRT-PCR. GAPDH was used as an internal reference gene. (F) Representative Western blot images showing the expression levels of RUNX2, COL1A1, OCN, and β-catenin in each group. GAPDH was used as a loading control. (G) Quantitative analysis of Western blot results (gray value ratio of target protein to GAPDH) in each group. All data are presented as mean ± SD, with statistical significance determined by unpaired two-tailed Student’s t-test (*p < 0.05; **p < 0.01; ***p < 0.001).
SSCs Alleviate Radiation-Induced Bone Injury in Mice
To assess the therapeutic efficacy of SSCs in mitigating radiation-induced bone loss, micro-computed tomography (micro-CT) analysis was performed. Representative 3D reconstructions (Figure 5A) revealed that IR caused severe deterioration of bone microarchitecture, which was notably ameliorated by SSC intervention at both 2- and 4-weeks post-irradiation. Quantitative morphometric analysis confirmed that IR significantly reduced bone mineral density (BMD; Figure 5B) and bone volume fraction (BV/TV; Figure 5C) relative to the control group. Conversely, the IR+SSC group maintained significantly higher BMD and BV/TV values at both time points. Regarding trabecular microarchitecture, IR exposure resulted in diminished trabecular thickness (Tb.Th; Figure 5D), reduced trabecular number (Tb.N; Figure 5E), and lower connectivity density (Conn.D; Figure 5F), alongside an expansion in trabecular separation (Tb.Sp; Figure 5G). Notably, SSC treatment rescued these trabecular parameters, yielding increased Tb.Th, Tb.N, and Conn.D, while concurrently reducing Tb.Sp compared to the IR-only group. Histological examination via H&E staining demonstrated that irradiation triggered profound marrow adiposity, which was effectively attenuated by SSC administration (Figure 5H and 5I). Furthermore, TRAP staining revealed that the IR-induced expansion of the osteoclast population was significantly blunted in the presence of SSCs (Figure 5J and 5K), suggesting that SSCs inhibit pathologically accelerated bone resorption. To further elucidate the molecular changes in the bone niche, we quantified the spatial expression of Osterix and β-catenin. Irradiation resulted in a profound loss of Osterix-positive cells, an effect that was significantly reversed by SSC intervention (Figure 5L and 5M). Furthermore, the expression of β-catenin, which was suppressed by IR, was significantly upregulated in the presence of SSCs (Figure 5N and 5O), indicating that SSCs counteract radiation-induced damage via the Wnt/β-catenin signaling axis. Taken together, our in vivo data establish that SSCs serve as a multifaceted therapeutic agent, capable of inhibiting pathological bone resorption and steatosis while simultaneously restoring the osteogenic potential of the irradiated skeleton. SSCs alleviate the radiation-induced bone injury in mice. (A–G) Micro-CT analysis of bone microstructure. (A) Representative micro-CT images of femurs. Quantitative analysis of (B) bone mineral density (BMD), (C) bone volume fraction (BV/TV), (D) trabecular thickness (Tb.Th), (E) trabecular number (Tb.N), (F) connectivity density (Conn.D), and (G) trabecular separation (Tb.Sp) at 2- and 4-weeks post irradiation. (H–K) Histological analysis (Scale bar: 100 μm). (H) H&E staining showing steatosis (arrows) and (I) quantitative analysis of steatotic lesions per field. (J) TRAP staining showing osteoclasts (arrows) and (K) quantitative analysis of osteoclast number per field. (L–O) Immunohistochemical staining of osteogenic markers (Scale bar: 100 μm). (L) Osterix staining and (M) quantitative analysis of Osterix-positive area. (N) β-catenin staining and (O) quantitative analysis of β-catenin-positive area. All experiments were conducted in three groups: Control, irradiation (IR), and IR plus SSC (IR+SSC) at 2- and 4-weeks post-irradiation. All data are presented as mean ± SD, with statistical significance determined by unpaired two-tailed Student’s t-test (*p < 0.05; **p < 0.01; ***p < 0.001)
Discussion
Radiation-induced bone damage represents a formidable clinical challenge, with functional exhaustion of osteogenic precursor cells (OPCs) serving as a central pathological hallmark. This study provides the first evidence that skeletal stem cells (SSCs) can rescue the proliferative and osteogenic capacity of irradiated OPCs by activating the Wnt/β-catenin signaling axis, offering a novel therapeutic strategy for skeletal radiotherapy sequelae. Our findings confirm that 8 Gy X-ray irradiation severely compromises OPC function, aligning with previous studies demonstrating that high-dose ionizing radiation (IR) suppresses the proliferation and differentiation of mesenchymal stem cells and osteoprogenitors. 22 Notably, we observed that IR downregulates β-catenin expression, suggesting that inhibition of this pathway is a primary driver of radiation-induced OPC dysfunction. This is consistent with evidence that high-dose radiation disrupts Wnt/β-catenin signaling in hematopoietic stem cells, thereby impairing their regenerative potential.23,24 Conversely, low-dose IR has been shown to activate this pathway and promote osteoclastogenesis, indicating that the effects of radiation on Wnt/β-catenin signaling are inherently dose-dependent. 25
The regenerative potential of SSCs is often attributed to their ability to orchestrate the osteogenic niche through paracrine signaling. 26 Our data demonstrate that SSC co-culture significantly mitigates apoptotic signaling and restores functional capacity in radiation-injured OPCs, pointing to a secretome-mediated rescue mechanism. Given that MSCs are known to secrete potent Wnt ligands (e.g., Wnt1, Wnt3a, Wnt5a) that induce β-catenin translocation in target cells,27,28 our observation of upregulated β-catenin in the IR+SSC group provides a plausible molecular basis for this recovery. Thus, SSCs likely serve as a paracrine source of Wnt ligands, effectively rebooting the Wnt/β-catenin pathway in the radiation-impaired skeletal microenvironment.
The mechanistic requirement of Wnt/β-catenin signaling was validated using MSAB to induce the targeted degradation of β-catenin. 29 We found that MSAB treatment abrogated the pro-osteogenic benefits conferred by SSC co-culture, effectively suppressing the expression of RUNX2, COL1A1, and OCN. These data establish that β-catenin stability is a prerequisite for the SSC-mediated rescue of radiation-injured OPCs. Our results are consistent with previous studies on skeletal repair, which demonstrate that Wnt/β-catenin signaling acts as a critical molecular switch governing the differentiation of SSCs into functional osteoblasts during bone regeneration.30,31
Despite the significance of our findings, this study has several limitations. First, we utilized a single high-dose radiation model, which may not fully recapitulate the fractionated radiotherapy protocols used in clinical oncology. Future research should evaluate the efficacy of SSCs in fractionated radiation models to better reflect patient treatment regimens. Second, while we identified the Wnt/β-catenin pathway as a key mediator, the specific paracrine factors secreted by SSCs that mediate this pathway activation remain to be characterized. Identifying these bioactive molecules will be crucial for developing cell-free targeted therapies. Furthermore, the optimal dosage and temporal window for SSC transplantation require further investigation to facilitate successful clinical translation.
Conclusion
In conclusion, our study demonstrates that SSCs effectively rescue the proliferation and osteogenic capacity of irradiated OPCs via activation of the Wnt/β-catenin signaling pathway. These findings elucidate a novel cellular mechanism underlying skeletal repair and provide a robust theoretical foundation for the clinical translation of SSC-based therapies in the treatment of radiation-induced bone injury.
Supplemental Material
Supplemental material - Skeletal Stem Cells Rescue Radiation-Induced Osteogenic Precursor Cell Dysfunction via the Wnt/β-Catenin Signaling Pathway
Supplemental material for Skeletal Stem Cells Rescue Radiation-Induced Osteogenic Precursor Cell Dysfunction via the Wnt/β-Catenin Signaling Pathway by Huahui Bian, Dongyang Zhao, Haoyu Wang, Hao Huang, Youyou Wang, Weibo Chen, Chang Liu, Yinyin Yang, Anqing Wu and Yulong Liu in Dose-Response.
Footnotes
Acknowledgements
The authors thank all the staff of Key Laboratory of Radiation Damage and Treatment of Jiangsu Provincial Universities and Colleges for expert technical assistance and administrative support.
Ethical Considerations
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of Soochow University (Ethics Approval Number: SUDA202110A0172; approved on October 9, 2021). All experiments were conducted in accordance with the ARRIVE guidelines for reporting animal research.
Author Contributions
Huahui Bian and Dongyang Zhao: Methodology, Data Curation, Visualization, Investigation, and Writing – Original Draft. Haoyu Wang, Hao Huang, Youyou Wang, Weibo Chen, Chang Liu, and Yinyin Yang: Data Curation, Visualization, and Investigation. Huahui Bian, Anqing Wu, and Yulong Liu: Project Administration, Funding Acquisition, Resources, and Writing – Review & Editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (No. U2267220, 81903248, and 12205215), the Gusu Talent Program (No. GSWS2022042), an Open Research Project of the State Key Laboratory of Radiation Medicine and Protection (No. GZK1202115, GZK12020042), and a Nuclear Medicine Research Project of the Discipline Construction Support Project (Phase II) of the General Hospital of Nuclear Industry (Nuclear Emergency Treatment, No. XKTJ-HTD2025002).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets utilized and/or examined in this study can be obtained from the corresponding author upon reasonable request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.