
Abstract
Radiotherapy is mainly a traditional treatment for breast cancer; however, the key genes and pathways in breast cancer associated with irradiation are not clear. In this study, we aimed to explore the messenger RNA expression changes between preradiation and postradiation breast cancer. The gene expression data set (GSE59733) was downloaded from Gene Expression Omnibus database. According to |log2FC (fold change) | ≥ 1 and with false discovery rate adjusted P value <.05, differentially expressed genes (DEGs) were screened and annotated by R programming software. The protein–protein interaction (PPI) network was conducted through STRING database, and subnetworks and hub genes were extracted by plug-in in Cytoscape. A total of 82 DEGs (74 upregulated and 8 downregulated genes) were identified. These DEGs mainly enriched in an intrinsic apoptotic signaling pathway and G-protein-coupled receptor binding. What’s more, tumor necrosis factor signaling pathway and interleukin 17 signaling pathway abnormally activated in postradiation tumor samples. Two characteristic subnetworks and 3 hub genes (FOS, CCL2, and CXCL12) were strongly distinguished in PPI network. Moreover, the expression level of the hub genes was confirmed in irradiated MCF-7 cell and SUM-159 cell using quantitative real-time polymerase chain reaction assay. These findings imply that these hub genes may play momentous function in breast cancer to irradiation.
Introduction
Breast cancer is one of the most prevalent cancers in women worldwide. 1,2 At present, radiotherapy has made remarkable achievements for solid tumor therapy, which especially acts as a positive role for most breast cancer. 3,4 For example, postoperative radiotherapy can effectively reduce the mortality and recurrence of breast cancer. 5,6 However, there is accumulating evidence suggesting that patients with breast cancer exhibit heterogeneous response to radiotherapy, seriously affecting the clinical efficacy and quality of life. 7 Previous research studies have shown that there are beneficial response to radiotherapy in patients with breast cancer characterized by Rec-positive or HER2-negative diagnosis. 8 Of note, the classification standard based on subtype is unable to fully describe clinically significant heterogeneity to radiotherapy among breast tumors. Therefore, it remains an urgent clinical problem to explore the difference in response to radiotherapy in breast cancer, which may effectively avoid overtreatment and undertreatment.
Recent studies suggested that abnormal gene expression was the principal contributor to cell behavior response to radiation. For example, Tob1 markedly mediated radiosensitivity in breast cancer cell MDA-MB-231 by targeting the JNK and p38 pathways. 9 Radiation promotes the migratory and invasive behavior in lung cancer cell A549 through transforming growth factor β–mediated epithelial–mesenchymal transition. 10 Furthermore, radiation induced the upregulated expression of Sox2 and Oct3/4, which is responsible for spherogenesis in cancer cells. 11 Interestingly, a recent study identified that vascular endothelial growth factor A and other factors secreted from irradiated MCF-7 cells contribute to angiogenic responses in endothelial cells. 12 However, the exact molecular mechanism of cell behavior response to radiation regrettably hasn’t been well researched at present. Thus, we boldly speculated that there might be some undiscovered gene that plays a key role in mediating the breast cancer cell behavior to radiotherapy.
Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) is the largest and most comprehensive database of public gene expression, providing a large amount of high throughput data for gene research. 13 It has been making a research hot spot to screen gene expression profiling to satisfy personalized breast cancer treatment for several years. For example, Pajic et al screened 11 potential microRNAs associated with local relapse following radiotherapy by gene expression profile and further confirmed that miR-139-5p may be an effective therapeutic target involved in DNA damage response to radiotherapy in breast cancer. 4 We screened the data set related to radiotherapy for breast cancer at GEO. The GSE59733 was selected for our research. The reason for choosing GSE59733 is that the treatment condition of GSE59733 is simple and clear, only before and after irradiation. There are few interference factors which are helpful to explain the influence of radiotherapy on breast cancer more clearly.
In this study, we identified DEGs with a cutoff degree of 2-fold change by microarray data analysis. We analyzed total transcriptional changes through functional annotation of DEGs. The hub genes, which are the possession of the complex interactions with other protein nodes, and primary subnetworks were extracted from the protein–protein interaction (PPI) network. Meanwhile, we exactly validated the hub gene expression trend in irradiated breast cancer cells by quantitative real-time polymerase chain reaction (qRT-PCR). Taken together, these findings provide new insights into cell response to radiation to further understand the important roles of messenger RNA (mRNA) differential expression in cellular radiation response.
Materials and Methods
Data Collection and Preprocessing
The raw expression matrix file from GSE59733 microarray data set was downloaded from GEO database supported by National Center of Biotechnology Information. It consists of 19 samples with early-stage breast cancer that received preoperative radiotherapy, including 9 preradiation tumor samples and 10 postradiation tumor samples. According to the annotation information provided by platform GPL8990 [Xcel] Affymetrix Human Almac Xcel Array, probe ID and Gene Symbol achieved transformation in downloaded expression matrix. The unrecognized probes were removed, and the repeated gene probes were merged by averaging using basic functions in R (version 3.5.1, https://www.r-project.org/). Missing data were imputed using the k-nearest neighbor algorithm by impute package 14 in R. The data normalization was also performed by normalizeBetweenArrays function in limma package 15 in R.
Identification and Functional Annotation of DEGs
The DEGs in postradiation tumor samples, compared to preradiation tumor samples, were screened from preprocessed gene expression data by limma package. The t test method was applied to calculate the adjusted P value of the DEGs. The adjusted P value <.05 and |log2FC (fold change)| ≥ 1 were selected as the cutoff criteria. To better describe the biological functions for DEGs involved in encoding protein, Gene Ontology (GO) functions analysis 16 and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis 17 were performed by clusterProfiler package 18 in R. The P value <.01 and Q value <.05 were used to restrict the output results for functional annotation.
The PPI Network Construction
STRING 19 (version10.5, https://string-db.org/) database is mainly used to describe the complex biological relationship between genes, containing a large amount of known and predicted PPI information. In this study, the DEGs involved in encoding proteins were retrieved in STRING database, and combined score ≥0.4 was identified as a major selection condition. Then, the PPI network was visualized by Cytoscape software. 20 Meanwhile, 2 typical network topological parameters were utilized to filter out hub nodes in PPI network by plugin cytoHubba, 21 in Cytoscape, including degree centrality 22 and betweenness centrality. 23 Subsequently, the plugin Molecular Complex Detection (MCODE) was applied to screening the primary modules of PPI network with the threshold set as follows: cutoff criteria degree = 2, K-core = 2, node score = 0.2, max depth = 100, and node number ≥5. What’s more, the extracted modules were analyzed by KEGG pathway analysis based on STRING database.
The MRNA Expression of Hub Genes in Normal and Tumor Samples
The UALCAN database 24 (http://ualcan.path.uab.edu/) is available and is a powerful database. It contains amounts of biological information regarding patients with cancer, such as survival information based on gene expression and its level. It also may help users to obtain more cancer transcriptome data and identify potential biomarkers. In this study, it was applied to evaluating mRNA expression data of hub genes between normal samples and tumor samples.
Cell Culture and Radiation
The human normal breast cell MCF-10A and the human breast cancer cell lines MCF-7, ZR-75-1, MDA-MB-231, SUM-159, and BT474 were obtained from American Type Culture Collection in the United States. The cells were cultured in Dulbecco’s Modified Eagle’s medium or RPMI medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin in 5% CO2 at 37 °C. Besides, the MCF-7 or SUM-159 cells (1 × 106) were seeded in 6-well plates. After 24 hours, MCF-7 or SUM-159 cells were exposed to 5 Gy irradiation dose using a 137Cs-ray source at a dose rate of 0.99 Gy/min.
The QRT-PCR Assay
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Reverse transcription was performed using a PrimeScript RT Reagent Kit (TaKaRa), according to the manufacturer’s instructions. SYBR Green Master (ROX) (Roche) was used to quantify hub genes in mRNA level, including FOS, CCL2, and CXCL12. Glyceraldehyde 3-phosphate dehydrogenase was amplified as control. The relative mRNA expression levels of hub genes were calculated using the 2−ΔΔCT method. The qRT-PCR cycles were as follows: 50 °C for 2 minutes, 95 °C for 10 minutes, and then 40 cycles of 95 °C for 15 seconds and 60 °C for 2 minutes. The primers are listed in Table 1. Each experiment was repeated at least 3 times.
List of Primers in this Study.

Statistical Analysis
The microarray data were analyzed by R software (version 3.5.1). Data in the present study were analyzed using SPSS statistical software (version 19.0). Statistical significance was assessed by comparing mean ± SD using a Student t test for independent groups. A 2-tailed P < .05 was considered statistically significant.
Results
Identification of DEGs in Breast Cancer After Irradiation
After data preprocessing, a total of 12 487 genes were mapped to probes. The normalization result for gene expression data was visualized with box plot (Figure S1). The result showed that the median values of each set of data were consistent, and the nonexperimental error was eliminated after normalization. According to the selected criteria (adjusted P value <.05 and |log2FC (fold change)| ≥ 1), a total of 82 DEGs, including 74 upregulated genes and 8 downregulated genes, were obtained after data processing in postradiation tumor samples, compared to the preradiation tumor samples. These DEGs are shown in Table S1. The volcano plot was applied to distribution for DEGs (Figure S2).
Gene Ontology and KEGG Analysis of DEGs
To explore the functional roles of the DEGs, we analyzed these candidate genes and found that a total of 62 GO terms were eligible for screening conditions (P value <.01 and Q value <.05) by clusterProfiler package in R programming. Fifty-seven GO terms were enriched in biological process (BP), and 5 GO terms were enriched in molecular function (MF). The GO enrichment analysis for BP ontology showed that these DEGs were mainly enriched in response to extracellular stimulus, nutrient levels, and intrinsic apoptotic signaling pathway in response to DNA damage (Figure 1A). In MF ontology, these DEGs were closely associated with G-protein-coupled receptor binding, chemokine receptor binding, and CXCR chemokine receptor binding (Figure 1B). Additionally, the results of KEGG pathway analysis revealed that these DEGs were involved in interleukin 17 signaling pathway and tumor necrosis factor (TNF) signaling pathway. This result is shown in Table 2.
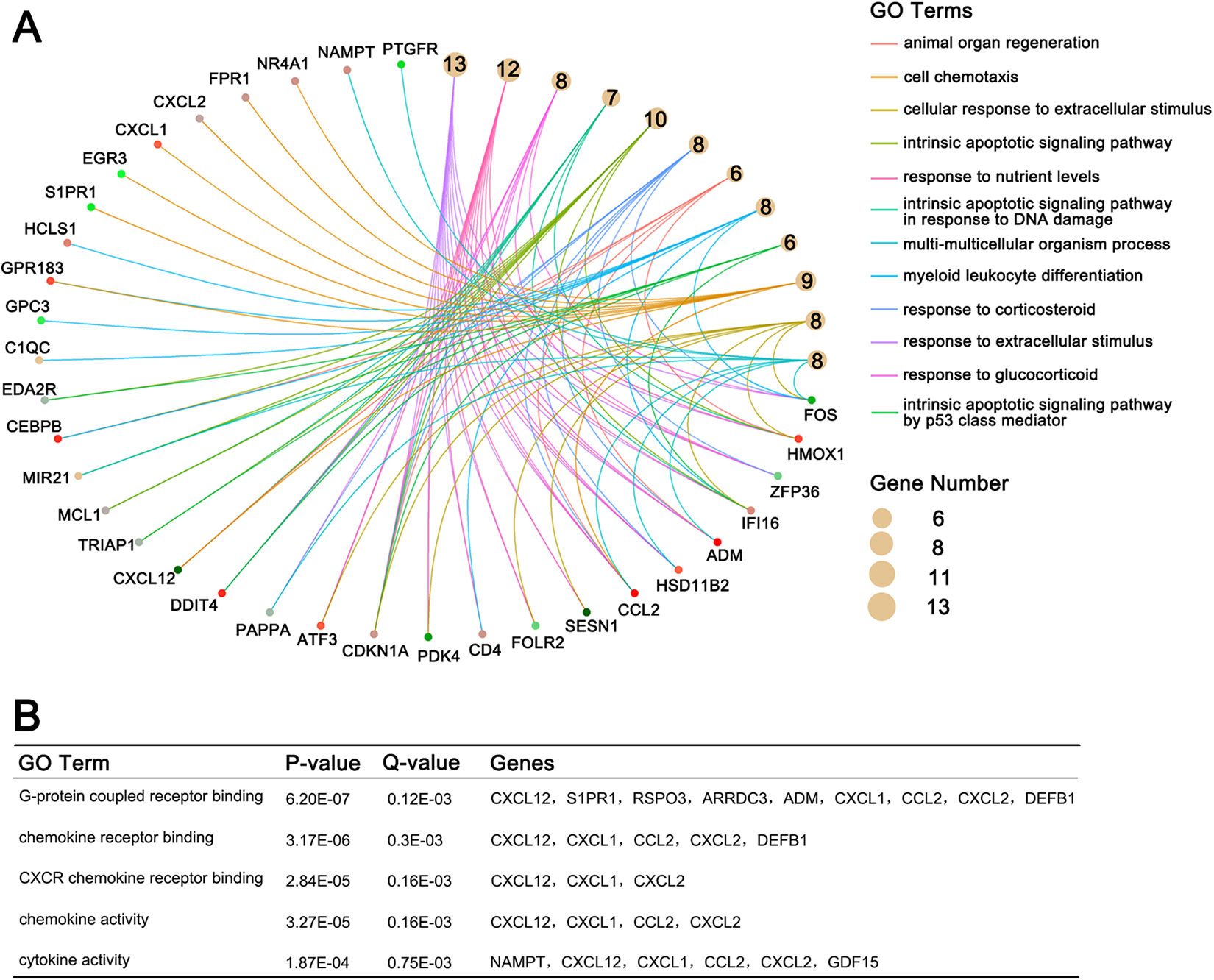
The results of Gene Ontology (GO) analysis. A, The top 12 GO terms in biological process. B, The top 5 GO terms in molecular function.
The Results of Kyoto Encyclopedia of Genes and Genomes Pathway Analysis.

The PPI Network Analysis of DEGs
To further explore the potential biological functions of DEGs, we constructed the PPI network by retrieving DEGs in STRING public database. The results showed that there were 73 nodes and 101 interactions in this network (Figure 2A). Top 6 hub genes were determined by calculating degree centrality and betweenness centrality, respectively. This result is shown in Table 3. The results suggested that a total of 3 hub genes were obtained by overlapping, including FOS, CCL2, and CXCL12. What’s more, 2 representative network modules were extracted from PPI network by MCODE plugin in Cytoscape 3.6.0. And the results of KEGG pathway analysis for each module suggested that these DEGs were highly involved in inflammation-associated signaling pathways, where DEGs in 2 modules are enriched in TNF signaling pathway, respectively (Figure 2B and C).

Construction and analysis of the protein–protein interaction (PPI) networks. A, The PPI networks. B, Module 1 and enriched pathways. C, Module 2 and enriched pathways.
The Hub Genes Obtained by Cytohubba.

Validation of the mRNA Expression of Hub Genes
Next, we found that the 3 hub genes (FOS, CCL2, and CXCL12) were downregulated in tumor samples, compared to normal samples based on UALCAN database (Figure 3A). Similarly, these results were confirmed in vitro. The mRNA expression level of the 3 hub genes in breast cancer cell lines were downregulated compared to normal breast cell MCF-10A (Figure 3B). To further verify the accuracy of microarray data analysis and explore the 3 hub genes response to radiation, we detected the relative mRNA expression trend of hub genes using qRT-PCR assay in irradiated breast cancer cells MCF-7 and SUM-159. As expected, our data demonstrated that the mRNA expression of these 3 hub genes were significantly increased after γ-irradiation (5 Gy) in 2 breast cancer cells which were consistent with results of data analysis (Figure 3C).
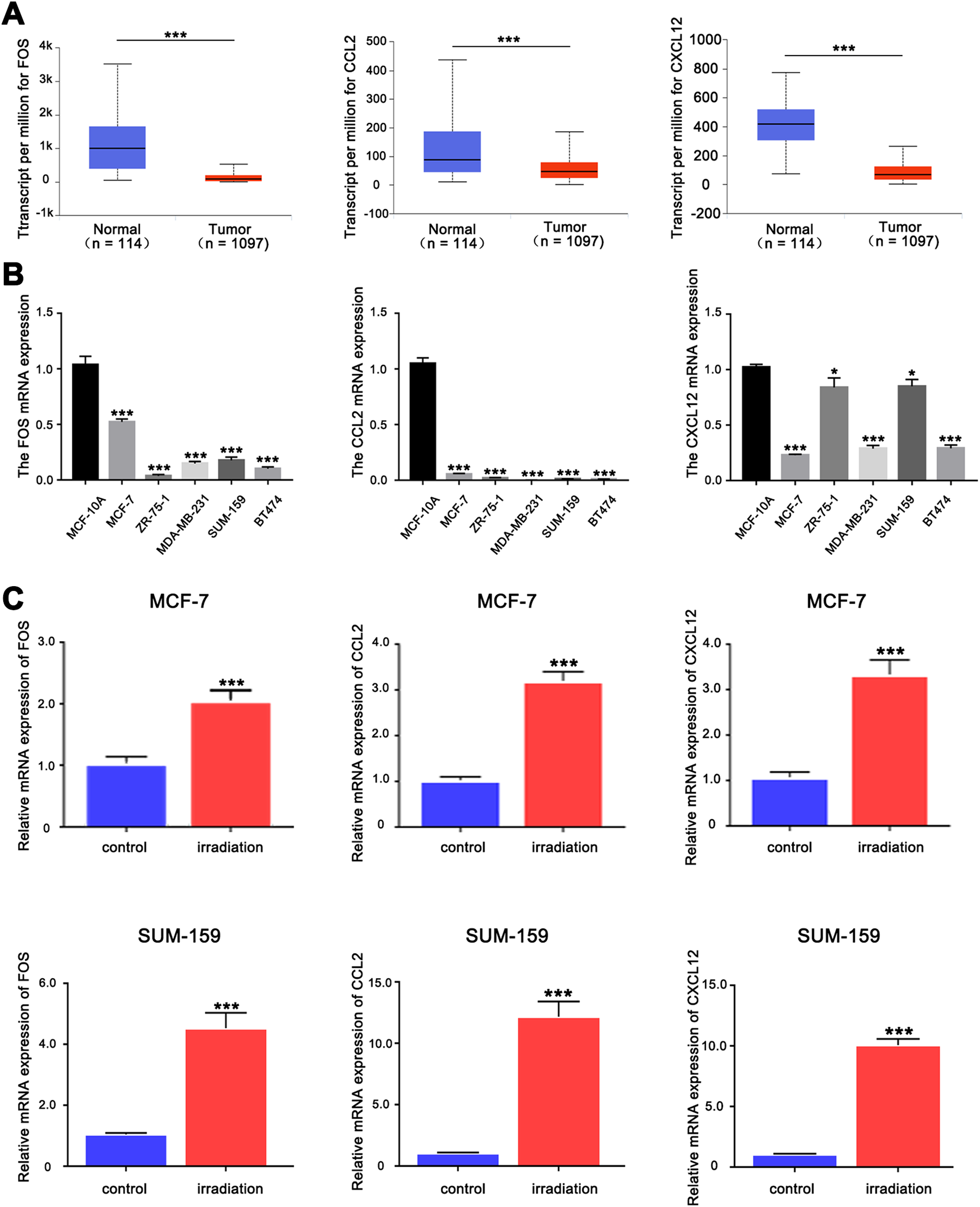
The expression levels of the 3 hub genes. A, The messenger mRNA (mRNA) expression data of the 3 hub genes were obtained from UALCAN database. B, The mRNA expression levels of the 3 hub genes in normal breast cell and breast cancer cell lines by quantitative real-time polymerase chain reaction (qRT-PCR). C, The mRNA expression levels of the 3 hub genes in irradiated MCF-7 and SUM-159 cells by qRT-PCR. ***P < .001.
Discussion
In this study, we retrieved gene expression profiling in response to radiation treatment in breast cancer from GEO database. It is found that the hidden networks of interaction regarding radiation-associated genes in GSE59733 data set haven’t yet been fully explored using bioinformatics. Interestingly, we identified 82 DEGs, including 74 upregulated genes and 8 downregulated genes, based on 19 tumor samples using algorithm in limma package. Moreover, the results from GO analysis and KEGG pathway analysis implied that these DEGs were mainly involved in extracellular stimulus, cell apoptosis, inflammation, immune responses, and DNA damage response. It has been evidenced that these BPs are closely related to radiation-induced response. 25 -28
Three outstanding hub proteins were filtered out from the PPI network, including FOS (AP-1 transcription factor), CCL2 (C-C motif chemokine ligand 2), and CXCL12 (C-XC motif chemokine ligand 12). And we validated the mRNA expression changes of FOS, CCL2, and CXCL12 in breast cancer cells after radiation. The results showed that the expression trend of these 3 genes was consistent with gene expression profiling. It implied that the 3 hub genes are closely associated with radiation-induced response through mediating a series of cascade reaction.
FOS is one of 3 hub genes which plays a critical role in the development and progression of breast cancer by mediating the transcription of AP-1 target genes. 29,30 However, there are fewer studies to uncover FOS response to radiation in breast cancer. Recent studies indicated that silencing FOS could sensitize glioma cells to radiation by disturbing DNA damage repair and cell cycle arrest. 26 In our analysis, FOS was one of the hub proteins with the highest interaction in the PPI network. Thus, whether altering its gene expression may affect the response to radiation of breast cancer cells is still an urgent need of in-depth study.
Additionally, CCL2 and CXCL12 as chemokines were mainly involved in immunoregulatory and inflammatory processes. In in vivo experiment, it has been reported that the expression of CCL2 and CXCl2 was significantly increased after radiation. 31,32 In this study, the results of functional annotation for DEGs further strengthen the evidence that the signaling pathways associated with inflammation are abnormally activated such as chemokine signaling pathway and TNF signaling pathway. There are studies pointing out the binding of chemokines and its ligand is involved in the biological responses of tumors to radiation. For example, CXCR4 and its receptor CXCL12 may regulate the survival of prostate cancer cells after irradiation. 33 And Kalbasi et al demonstrated chemokine CCL2 could prevent the recruitment of inflammatory monocytes to tumors and reduce the growth of tumors when combined with radiotherapy. 34
In conclusion, the 3 hub genes obtained from the PPI network are closely associated with radiation response, which may be expected to be therapeutic targets or diagnostic markers for radiotherapy patients in the future. Nevertheless, it still required a rigorous experiment to support in the following research. Anyway, this study provided a new insight into breast cancer response to radiation.
Supplemental Material
Supplemental Material, Figure_S1 - Identification of Key Genes and Pathways Associated With Irradiation in Breast Cancer Tissue and Breast Cancer Cell Lines
Supplemental Material, Figure_S1 for Identification of Key Genes and Pathways Associated With Irradiation in Breast Cancer Tissue and Breast Cancer Cell Lines by Changchun Zhu, Chang Ge, Junbo He, Xueying Zhang, Guoxing Feng and Saijun Fan in Dose-Response
Supplemental Material
Supplemental Material, Figure_S2 - Identification of Key Genes and Pathways Associated With Irradiation in Breast Cancer Tissue and Breast Cancer Cell Lines
Supplemental Material, Figure_S2 for Identification of Key Genes and Pathways Associated With Irradiation in Breast Cancer Tissue and Breast Cancer Cell Lines by Changchun Zhu, Chang Ge, Junbo He, Xueying Zhang, Guoxing Feng and Saijun Fan in Dose-Response
Supplemental Material
Supplemental Material, Table_S1._the_differentially_expressed_genes. - Identification of Key Genes and Pathways Associated With Irradiation in Breast Cancer Tissue and Breast Cancer Cell Lines
Supplemental Material, Table_S1._the_differentially_expressed_genes. for Identification of Key Genes and Pathways Associated With Irradiation in Breast Cancer Tissue and Breast Cancer Cell Lines by Changchun Zhu, Chang Ge, Junbo He, Xueying Zhang, Guoxing Feng and Saijun Fan in Dose-Response
Footnotes
Authors’ Note
Changchun Zhu and Chang Ge equally contributed to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China [grant number 81703042, 81572969 and 81730086] and CAMS Innovation Fund for Medical Sciences [CIFMS, 2017-I2M-1-016], and this project was funded by China Postdoctoral Science Foundation [grant number 2018T110071].
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.