
Abstract
This study aims to examine the effect of linolenic acid on the vasodilation or vasoconstriction induced by acetylcholine and bupivacaine in isolated rat aortae and its underlying mechanism. The effect of linolenic acid on the vasodilation induced by acetylcholine, the calcium ionophore A23187, sodium nitroprusside, and 8-bromoguanosine 3′,5′-cyclic monophosphate sodium salt (bromo-cyclic guanosine monophosphate [bromo-cGMP]) in endothelium-intact and endothelium-denuded aortae was examined. Linolenic acid inhibited vasodilation induced by acetylcholine, calcium ionophore A23187, and sodium nitroprusside. However, this fatty acid increased bromo-cGMP-induced vasodilation in endothelium-denuded aortae. Linolenic acid increased bupivacaine-induced contraction in endothelium-intact aortae, whereas it decreased bupivacaine-induced contraction in endothelium-intact aortae with Nω-nitro-
Keywords
Introduction
Lipid emulsion is effective in treating local anesthetic systemic toxicity. 1 In addition, lipid emulsion as a nonspecific antidote has been reported to be effective in alleviating cardiovascular collapse induced by toxic doses of highly lipid soluble drugs (log [octanol/water partition coefficient]: >2.0), including verapamil, amlodipine, and diphenhydramine. 1,2 Intralipid, whose fatty acid component consists of 100% long-chain fatty acids, attenuates acetylcholine-induced nitric oxide (NO)-mediated relaxation. 3 Furthermore, Intralipid increases left ventricular systolic pressure via the inhibition of NO. 4 In addition, administration of lipid emulsions causes increased blood pressure and systemic vascular resistance. 5 -8 Intralipid, which is derived from soybean oil, contains 53% linoleic acid, 24% oleic acid, 11% palmitic acid, 8% linolenic acid, and 4% stearic acid. 9 Both linoleic acid (C18:2n-6) and linolenic acid (C18:3n-3) among long-chain fatty acids contained in Intralipid are essential fatty acids, which should be supplied by diet, and polyunsaturated long-chain fatty acids. 9 Free fatty acids, including stearic acid, decrease acetylcholine-induced NO-mediated relaxation. 10 Moreover, a toxic concentration of aminoamide local anesthetics induces vasodilation (attenuated vasoconstriction) partially mediated by endothelial NO release, whereas a lipid emulsion containing linolenic and linoleic acids inhibits vasodilation induced by a toxic concentration of levobupivacaine, which seems to be partially associated with the inhibition of endothelial NO release. 11 -15 On the other hand, α-linolenic acid improves the impaired acetylcholine-induced relaxation caused by cigarette smoking, suggesting that α-linolenic acid and its metabolite may induce NO production. 16,17 First, the goal of this study is to examine the effect of long-chain polyunsaturated essential fatty acids, linolenic, and linoleic acid on acetylcholine-induced NO-mediated relaxation and to elucidate the underlying cellular mechanism. Second, the effects of linolenic acid on the concentration–response (vasoconstriction and vasodilation) curves induced by bupivacaine in isolated rat aortae and its underlying mechanism with a particular focus on endothelial NO were examined.
Materials and Methods
The Institutional Animal Care and Use Committee of Gyeongsang National University approved all the experimental protocols (GNU-160414-R0019), and all the experimental procedures were performed in accordance with the Guideline of the Care and Use of Laboratory Animal of Gyeongsang National University.
Preparation of Isolated Rat Aortae for Isometric Tension Measurement
Isolated rat aortae for isometric tension measurement were prepared as described previously. 18,19 Carbon dioxide (100%) was used to euthanize male Sprague-Dawley (body weight: 250-300 g). The descending thoracic part of the aorta was dissected and removed from the thorax. Aortae were immersed in Krebs solution, which is composed of the following components (mM): sodium chloride (118), glucose (11), sodium bicarbonate (25), potassium chloride (4.7), calcium chloride (2.4), magnesium sulfate (1.2), and monopotassium phosphate (1.2). The periaortic tissues, such as fat and connective tissue, were removed under a microscope. The isolated aorta was cut into aortic rings 2.5 mm in length. The endothelium of some isolated descending thoracic aortae was removed by inserting two 25-gauge needles into the lumen of aortic rings and rolling the isolated aortic ring forward and backward. Isolated thoracic aortae were suspended in a Grass isometric transducer (FT-03, Grass Instrument, Quincy, Massachusetts) attached to an organ bath with 10 cc Krebs solution maintained at 37 C. Based on a previous similar experiment, a baseline resting tension of 3.0 g was chosen, and this baseline resting tension was maintained for 120 minutes to reach equilibrium. 20 The Krebs solution was replaced by fresh Krebs solution every 40 minutes. The pH at 7.4 of Krebs solution was maintained by aerating Krebs solution with 95% oxygen and 5% carbon dioxide. To confirm endothelial integrity, after phenylephrine (10−7 M) produced a sustained and stable contraction, acetylcholine (10−5 M) was added into the organ bath, and more than 85% relaxation from phenylephrine-induced contraction was considered to be endothelium-intact aortic rings. In addition, to verify endothelial denudation of isolated rat aortae, after phenylephrine (10−8 M) produced a sustained and stable contraction, acetylcholine (10−5 M) was added into the organ bath, and less than 15% acetylcholine-induced relaxation was regarded as endothelium-denuded aortic rings. The isolated endothelium-intact and endothelium-denuded aortic rings showing acetylcholine-induced relaxation were washed with fresh Krebs solution to restore baseline resting tension. Then, the contraction was induced by isotonic 60 mM KCl in some isolated aortic rings, and this contraction was used as a reference value to express the magnitude of contraction induced by dexmedetomidine and bupivacaine. Krebs solution with isotonic 60 mM KCl used to induce contraction was composed of sodium chloride (63.2 mM), potassium chloride (60 mM), sodium bicarbonate (25 mM), glucose (10 mM), calcium chloride (2.4 mM), magnesium sulfate (1.2 mM), and monopotassium phosphate (1.2 mM). Then, the isolated aortic rings with isotonic 60 mM KCl-induced contraction were washed with fresh Krebs solution to restore baseline resting tension. The following experimental protocols were performed.
Experimental Protocol
First, the effect of linolenic and linoleic acid (5 × 10−6 and 5 × 10−5 M) on the NO-induced vasodilation produced by endothelial muscarinic receptor-mediated vasodilator acetylcholine in endothelium-intact rat aortae was examined. Isolated endothelium-intact rat aortae were pretreated with linolenic and linoleic acid for 20 minutes. Then, phenylephrine (10−6 M) was added into the organ bath to produce vasoconstriction in the presence or absence of either linolenic or linoleic acid and then acetylcholine (10−8 to 10−5 M) was cumulatively added into the organ bath to produce vasodilation induced by acetylcholine.
Second, the effect of linolenic acid (5 × 10−5 M) on the vasodilation induced by endothelial receptor-independent vasodilator calcium ionophore A23187 and NO donor sodium nitroprusside in endothelium-intact and endothelium-denuded rat aortae was examined. Isolated endothelium-intact and endothelium-denuded rat aortae were pretreated with linolenic acid for 20 minutes. Then, 10−6 and 10−7 M phenylephrine was added to the organ bath to produce a sustained and stable vasoconstriction in endothelium-intact and endothelium-denuded rat aortae, respectively, in the presence or absence of linolenic acid. Then, calcium ionophore A23187 (10−9 to 10−6 M) and sodium nitroprusside (10−10 to 10−7 M) were cumulatively added into the organ bath with endothelium-intact and endothelium-denuded rat aortae, respectively, to produce vasodilation induced by calcium ionophore A23187 or sodium nitroprusside.
Third, the effect of GW1100, an inhibitor of G-protein–coupled receptor 40, which is known as free fatty acid receptor 1, and ethanol, which is used for the dissolution of linolenic acid, on acetylcholine-induced NO-mediated relaxation in endothelium-intact rat aortae with or without linolenic acid was examined. 21 The endothelium-intact rat aortae were pretreated with GW1100 (10−5 M) for 15 minutes followed by linolenic acid (5 × 10−5 M) for 20 minutes or ethanol (0.1%) and linolenic acid (5 × 10−5 M) alone for 20 minutes. Then, phenylephrine (10−6 M) was added into the organ bath to produce a sustained and stable contraction in the presence or absence of the combined treatment with GW1100 and linolenic acid, ethanol, or linolenic acid alone. Then, acetylcholine (10−8 to 10−5 M) was cumulatively added into the organ bath to produce vasodilation.
Fourth, the effect of linolenic acid on the vasodilation induced by cyclic guanosine monophosphate (cGMP) analog 8-bromoguanosine 3′,5′-cyclic monophosphate sodium salt (bromo-cGMP), nonspecific vasodilator papaverine, and calcium channel blocker diltiazem in endothelium-denuded rat aortae was examined. Isolated endothelium-denuded rat aortae were pretreated with linolenic acid (5 × 10−5 M) for 20 minutes. Then, phenylephrine (10−7 M) was added into the organ bath to produce a sustained and stable contraction in the presence or absence of linolenic acid. Bromo-cGMP (10−10 to 10−4 M), papaverine (10−7 to 3 × 10−5 M), and diltiazem (10−8 to 10−4 M) were cumulatively added into the organ bath to produce vasodilation.
Fifth, as the contraction induced by the α-2 adrenoceptor agonist dexmedetomidine is inhibited by endothelial NO, the effect of linolenic acid on the contraction induced by dexmedetomidine in endothelium-intact rat aortae was examined.
22
The isolated endothelium-intact rat aortae were pretreated with linolenic acid (5 × 10−5 M) for 20 minutes. Then, dexmedetomidine (10−9 to 10−6 M) was cumulatively added into the organ bath to produce a dexmedetomidine concentration–response curve in the endothelium-intact rat aortae with or without linolenic acid and endothelium-denuded rat aortae to confirm that dexmedetomidine-induced contraction is attenuated by endothelial NO.
22
After dexmedetomidine (10−6 M) produced maximal contraction in endothelium-intact rat aortae with or without linolenic acid, the nitric oxide synthase (NOS) inhibitor Nω-nitro-
Finally, the effect of extracellular calcium on the bupivacaine-induced contraction was examined. Isolated endothelium-denuded aortae were treated with calcium-free Krebs solution for 20 minutes. Bupivacaine (2 × 10−5 M) was added into the organ bath to produce vasoconstriction in the presence or absence of calcium-free Krebs solution. Then, calcium (1.25 and 2.5 mM) and linolenic acid (5 × 10−5 M) were added into the organ bath, showing bupivacaine-induced contraction with and without calcium-free Krebs solution, respectively. In addition, the effects of endothelial denudation and
Cell Culture
Human umbilical vein endothelial cells (EA.hy926 cells, American Type Culture Collection, Manassas, Virginia) were cultured in Endothelial Cell Medium (ScienCell Research Laboratories, Carlsbad, California) supplemented with 20% fetal bovine serum, 1% penicillin and streptomycin, and 1% endothelial cell growth supplement, as described previously. 23 Cells were plated onto gelatin (Difco, Becton Dickinson, New Jersey)-coated 100-mm culture dishes and incubated at 37°C in a humidified atmosphere containing 5% CO2 with media change every 2 days. Upon reaching confluence, the cells were dissociated with 0.025% trypsin–ethylenediaminetetraacetic acid solution and split at a 1:4 ratio. For our experiments, cells between passages 2 and 5 were seeded on to dishes (105 cells/100-mm dish) and cultured until they reached 70% confluence, followed by serum starvation overnight prior to drug treatment.
Western Blot Analysis
Western blot analysis was carried out as described previously. 23 Briefly, cells were lysed in the PRO-PREP protein extraction solution to isolate the total cell extract by centrifugation at 16 000×g for 15 minutes at 4°C. The protein concentrations were measured using the Bradford method. The protein samples to be loaded in the gel were prepared by mixing equal volumes of protein lysates with 2× sodium dodecyl sulfate sample buffer (0.1 M Tris–HCl, 20% glycerol, 4% sodium dodecyl sulfate, and 0.01% bromophenol blue). A total of 25 µg protein per sample was separated by 8% or 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis for 90 minutes at 110 V. The separated proteins were electrophoretically transferred to polyvinylidene difluoride membranes for 1 hour at 190 mA. Then, the membranes were blocked in Tris-buffered saline containing TWEEN 20 (TBST) with 5% (wt/vol) nonfat dried milk for 1 hour at room temperature and incubated overnight at 4°C with specific primary antibodies (anti-endothelial NOS [eNOS] and anti-phospho-eNOS: Cell Signaling Technology, Beverly, Massachusetts) diluted 1:1000 in TBST containing 5% (wt/vol) skim milk powder or 5% bovine serum albumin. After washing the membranes in TBST, bound antibodies were incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G diluted 1:5000 in TBST containing 5% (wt/vol) skim milk for 1 hour at room temperature. The membranes were washed in TBST, and the immunoreactive bands were detected by chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Thermo Scientific, Rockford, Illinois) using X-ray film ( SuperRX-N Fuji Medical X-ray Film, Japan) or ChemiDoc Touch Imaging System (Bio-Rad Laboratories Inc, Berkeley, California).
Cyclic GMP Measurement
Cyclic GMP measurement was performed as described previously.
24
The descending thoracic aortic strips were immersed in organ bath with 10 mL Krebs solution for 60 minutes. Endothelium-denuded thoracic aortic strips from the same rat aorta were untreated with drugs and treated with sodium nitroprusside (10−7 M) alone for 1 minute and linolenic acid (5 × 10−5 M) for 30 minutes followed by sodium nitroprusside (10−7 M) for 1 minute. Endothelium-intact thoracic aortic strips from the same rat aorta were untreated with drugs and treated with bupivacaine (3 × 10−4 M) alone for 1 minute or linolenic acid (5 × 10−5 M) alone for 31 minutes, linolenic acid (5 × 10−5 M) for 30 minutes followed by bupivacaine (3 × 10−4 M) for 1 minute, and
Materials
All the chemicals with the highest purity were obtained from commercially available companies. Linolenic and linoleic acid, phenylephrine, acetylcholine, calcium ionophore A23187, sodium nitroprusside, bromo-cGMP, papaverine, diltiazem, and
Statistical Analysis
Data are shown as the mean ± standard deviation. Data are expressed as the percentage of maximal contraction induced by phenylephrine or isotonic 60 mM KCl. The effects of linolenic and linoleic acid, ethanol, GW1100,
Results
Linolenic acid (5 × 10−6 and 5 × 10−5 M) inhibited acetylcholine-induced relaxation (Figure 1A;
P
< .05 vs control at 10−7, 3 × 10−6, and 10−5 M) in endothelium-intact rat aortae in a concentration-dependent manner, whereas a higher concentration of linoleic acid (5 × 10−5 M) inhibited acetylcholine-induced relaxation (Figure 1B;
P
< .05 vs control at 3 × 10−7 to 10−5 M) in endothelium-intact rat aortae. Linolenic acid (5 × 10−5 M) inhibited calcium ionophore A23187-induced vasodilation (Figure 2A;
P
< .01 vs control at 3 × 10−8 to 3 × 10−7 M) in endothelium-intact rat aortae. Calcium ionophore A23187 (3 × 10−7 M) produced maximal vasodilation in the control group with endothelium-intact aorta, and the highest concentration of calcium ionophore A23187 (10−6 M) produced vasoconstriction (Figure 2A;
P
< .001 vs 3 × 10−7 M). However, the highest concentration of calcium ionophore A23187 (10−6 M) produced maximal vasodilation in endothelium-intact aorta pretreated with linolenic acid (Figure 2A). In addition, linolenic acid (5 × 10−5 M) attenuated sodium nitroprusside-induced vasodilation in endothelium-denuded rat aortae (Figure 2B;
P
< .001 vs control at 3 × 10−9 to 3 × 10−8 M). The G-protein–coupled receptor 40 inhibitor GW1100 had no effect on acetylcholine-induced relaxation in endothelium-intact rat aortae treated with linolenic acid (5 × 10−5 M, Figure 3A). Ethanol (0.1%) had no effect on acetylcholine-induced relaxation in endothelium-intact rat aortae (Figure 3B). However, linolenic acid (5 × 10−5 M) increased vasodilation induced by the cGMP analog bromo-cGMP in endothelium-denuded rat aortae (Figure 4A;
P
< .05 vs control at 10−7 to 10−4 M). In addition, linolenic acid (5 × 10−5 M) enhanced vasodilation induced by nonspecific vasodilator papaverine (Figure 4B;
P
< .001 vs control at 10−5 M) and calcium channel blocker diltiazem (Figure 4C;
P
< .001 vs control at 10−6 to 10−4 M). Linolenic acid increased dexmedetomidine-induced contraction (Figure 5A and B;
P
< .01 vs control at 3 × 10−7 and 10−6 M) in endothelium-intact aortae. However, post-treatment with

Effect of linolenic acid (A) and linoleic acid (B) on acetylcholine-induced relaxation in isolated endothelium-intact rat aortae. Data (A: control, 5 × 10−6 and 5 × 10−5 M linolenic acid: N = 10, 5, and 6, respectively; B: control, 5 × 10−6 and 5 × 10−5 M linoleic acid: N = 6, 5, and 5, respectively) are shown as mean ± standard deviation and expressed as the percentage of maximal contraction induced by phenylephrine. N indicates the number of rats in which thoracic aortae were obtained. *P < .05 and † P < .001 versus control. # P < .001 versus 5 × 10− 6 M linolenic acid.

A, Effect of linolenic acid (N = 11) on the calcium ionophore A23187-induced relaxation in isolated endothelium-intact rat aortae. Data are shown as mean ± standard deviation and expressed as the percentage of maximal contraction induced by phenylephrine. N indicates the number of rat thoracic aortae. *P < .01 and † P < .001 versus control. # P < .001 versus 3 × 10− 7 M calcium ionophore A23187 in linolenic acid (5 × 10− 5 M). B, Effect of linolenic acid (N = 8) on sodium nitroprusside (SNP)-induced relaxation in isolated endothelium-denuded rat aortae. Data are shown as mean ± standard deviation and expressed as the percentage of maximal contraction induced by phenylephrine. N indicates the number of isolated rat aortae. *P < .001 versus control.

A, Effect of GW1100 on the acetylcholine-induced relaxation in isolated endothelium-intact rat aortae treated with linolenic acid. Data (N = 6) are shown as the mean ± standard deviation and expressed as the percentage of maximal contraction induced by phenylephrine. N indicates the number of isolated rat aortae. B, Effect of ethanol on the acetylcholine-induced relaxation in isolated endothelium-intact rat aortae. Data (N = 8) are shown as mean ± standard deviation and expressed as the percentage of maximal contraction induced by phenylephrine. N indicates the number of isolated rat aortae.

Effect of linolenic acid on the relaxation induced by 8-bromoguanosine 3′,5′-cyclic monophosphate sodium salt (bromo-cGMP; A: N = 8), papaverine (B; N = 8), and diltiazem (C; N = 8) in isolated endothelium-denuded rat aortae. Data are shown as mean ± standard deviation and expressed as the percentage of maximal contraction induced by phenylephrine. N indicates the number of isolated rat aortae. *P < .05, † P < .01, and # P < .001 versus control.

Concentration–response curves induced by cumulative addition of dexmedetomidine (A), followed by Nω-nitro-

Effect of linolenic acid on the bupivacaine concentration–response curves in endothelium-intact aortae without (A; N = 6) or with (B; N = 7) Nω-nitro-

A, Effect of linolenic acid (LA; 5 × 10−5 M) on the bupivacaine (BPV; 2 × 10−5 M)-induced contraction in the Krebs solution and effect of cumulative addition of calcium chloride (Ca; 1.25 and 2.5 mM) on the BPV (2 × 10−5 M)-induced contraction in the calcium-free Krebs solution. Data (N = 10) are shown as mean ± standard deviation and expressed as the percentage of maximal contraction induced by isotonic 60 mM KCl. N indicates the number of isolated rat aortae. *P < .001 versus BPV in the Krebs solution. †
P < .001 versus BPV in the Krebs solution or calcium-free Krebs solution. #
P < .001 versus Ca (1.25 mM) in the calcium-free Krebs solution. B, Effect of Nω-nitro-
Linolenic acid (5 × 10−6 and 5 × 10−5 M) and linoleic acid (5 × 10−5 M) inhibited acetylcholine (10−5 M)-induced eNOS phosphorylation (at Ser1177) in human umbilical vein endothelial cells (Figure 8A; P < .001 vs acetylcholine alone). Bupivacaine (3 × 10−4 M) induced eNOS phosphorylation (at Ser1177) in human umbilical vein endothelial cells (Figure 8B; P < .01 vs control). However, linolenic acid (5 × 10−5 M) inhibited bupivacaine (3 × 10−4 M)-induced eNOS phosphorylation (Figure 8B; P < .001 vs bupivacaine alone).

A, Effect of linolenic acid (LA) and linoleic acid on endothelial nitric oxide synthase (eNOS) phosphorylation (at Ser1177) induced by acetylcholine (ACH) in human umbilical vein endothelial cells (HUVECs). HUVECs were treated with ACH (10−5 M) alone for 4 minutes and pretreated with LA (5 × 10− 6 and 5 × 10− 5 M) and linoleic acid (5 × 10−5 M) for 1 hour, followed by ACH (10−5 M) for 4 minutes. Data (N = 3) are shown as mean ± standard deviation. N indicates the number of independent experiments. *P < .001 versus control. † P < .001 versus ACH alone. B, Effect of LA on eNOS phosphorylation (at Ser1177) induced by bupivacaine (BPV) in HUVECs. HUVECs were treated with BPV (3 × 10−4 M) alone for 10 minutes and pretreated with LA (5 × 10−5 M) for 1 hour, followed by BPV (3 × 10−4 M) for 10 minutes or LA (5 × 10−5 M) alone for 70 minutes. Data (N = 3) are shown as mean ± standard deviation. N indicates the number of independent experiments. *P < .01 and † P < .05 versus control. # P < .001 versus BPV alone. p-eNOS indicates phosphorylated endothelial nitric oxide synthase; t-eNOS: total endothelial NOS.
Linolenic acid (5 × 10−5 M) decreased sodium nitroprusside (10−7 M)-induced increased cGMP formation (P < .001 vs sodium nitroprusside; Figure 9A) in endothelium-denuded aortic strips. Bupivacaine (3 × 10−4 M) decreased cGMP formation (Figure 9B;
P
< .001 vs control) in endothelium-intact aortic strips. Pretreatment with linolenic acid (5 × 10−5 M) or
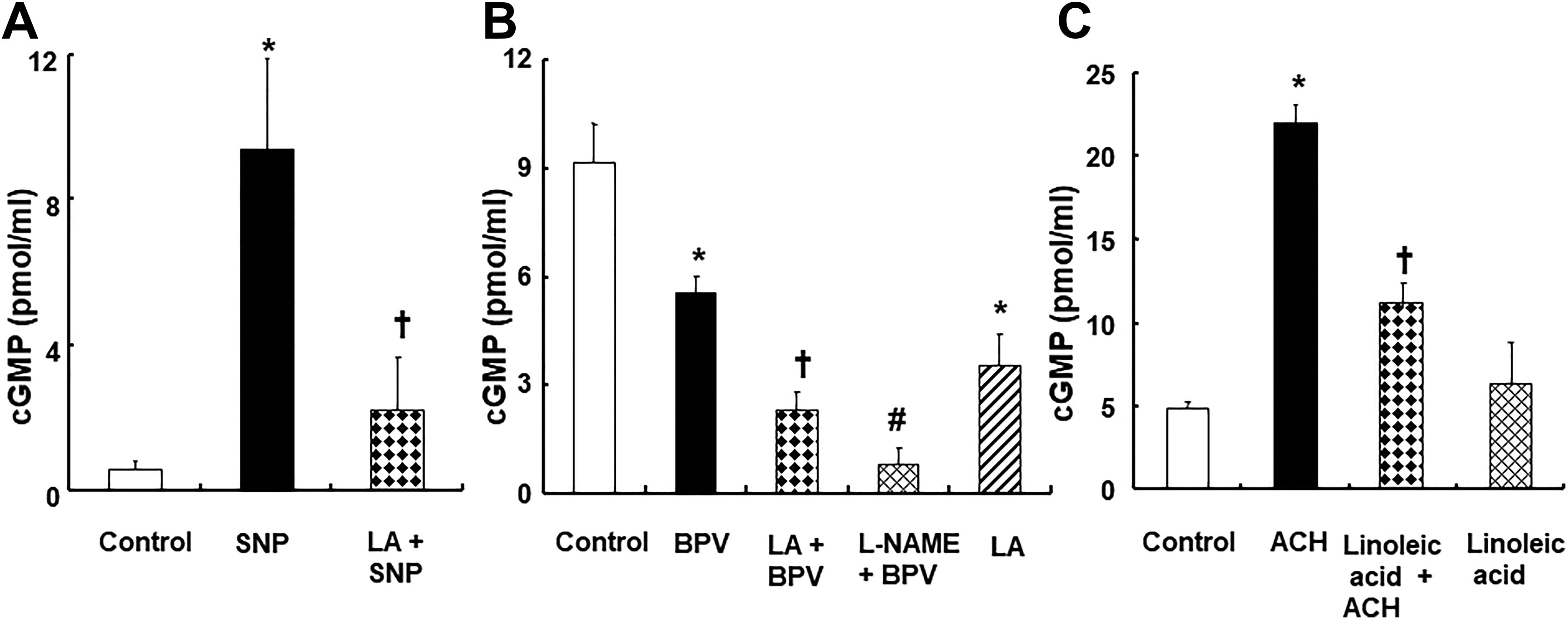
A, Effect of linolenic acid (LA, 5 × 10−5 M) on sodium nitroprusside (SNP, 10−7 M)-induced cyclic guanosine monophosphate (cGMP) formation in endothelium-denuded aortic strips. Data (N = 3) are shown as mean ± standard deviation. N indicates the number of rats. *P < .001 versus control. †
P < .001 versus SNP alone. B, Effect of LA (5 × 10−5 M) and Nω-nitro-
Discussion
This study suggests that linolenic acid inhibits acetylcholine-induced NO-mediated relaxation in endothelium-intact rat aortae via the inhibition of site just upstream of cGMP formation. The major findings of this study are as follows: (1) Linolenic acid more potently inhibited acetylcholine-induced relaxation than linoleic acid; (2) linolenic acid inhibited vasodilation induced by calcium ionophore A23187 and it did not produce the calcium ionophore A23187 (10−6 M)-induced vasoconstriction observed in the control group; (3) linolenic acid decreased the vasodilation induced by sodium nitroprusside; (4) linolenic acid attenuated vasodilation induced by a toxic concentration of bupivacaine (3 × 10−4 M) in endothelium-intact rat aortae (Figure 6A), whereas
Nitric oxide is produced from
Linolenic acid increased the vasodilation induced by nonspecific vasodilator papaverine, which produces vasodilation via both inhibition of phosphodiesterase to produce increased amounts of cGMP and cyclic adenosine monophosphate and inhibition of calcium influx. 30,31 In addition, linolenic acid enhanced vasodilation induced by diltiazem, which produces vasodilation via inhibition of calcium influx. 32 As polyunsaturated fatty acid inhibits voltage-dependent L-type calcium current, this enhancement of vasodilation induced by papaverine (Figure 4B) and diltiazem (Figure 4C) may be associated with linolenic acid’s inhibition of calcium channels. 33 Similar to these results, the highest concentration (10− 6 M) of calcium ionophore A23187 produced vasoconstriction in the control group, whereas it produced maximal vasodilation in linolenic acid group, which may be due to the inhibitory effect of linolenic acid on the calcium influx into vascular smooth muscle induced by calcium ionophore A23187 (10− 6 M). Moreover, similar to previous reports involving aminoamide local anesthetics, the calcium-free state nearly abolished bupivacaine-induced contraction and the subsequent addition of calcium increased bupivacaine-induced contraction (Figure 7A), suggesting that linolenic acid may inhibit calcium influx from the extracellular space. 11,12,34 Thus, further study regarding the effect of linolenic acid on the intracellular calcium level is needed. In contrast to the inhibitory effect of linolenic acid on NO-induced relaxation, linolenic acid-induced enhancing effects of vasodilation caused by papaverine and bromo-cGMP appear to be associated with the site below cGMP formation. Therefore, this linolenic acid-mediated inhibition of NO-induced vasodilation may be specific.
In agreement with previous reports using aminoamide local anesthetics, the vasodilation induced by a toxic concentration (3 × 10−4 M) of bupivacaine is mediated by NO (Figure 7B). 11 -13,18,35 In addition, a lipid emulsion containing linolenic acid reverses vasodilation induced by a toxic concentration of levobupivacaine in endothelium-intact aortae and reduces levobupivacaine-induced eNOS phosphorylation in human umbilical vein endothelial cells and rat aortic endothelium. 14,15 Taken together, the evidence suggests that linolenic acid-induced inhibition of vasodilation caused by NO observed in the current study appears to partially contribute to lipid emulsion-mediated inhibition of severe vasodilation (vascular collapse) caused by NO release induced by toxic concentrations of local anesthetics. 11 -15,18 In addition, as linolenic acid increased dexmedetomidine-induced contraction in endothelium-intact aortae, a patient concurrently taking Intralipid and dexmedetomidine for parenteral nutrition and sedation, respectively, in the intensive care unit may show slightly increased blood pressure compared with a patient taking only dexmedetomidine. However, this study has some limitations. First, the aorta was used for isometric tension measurement in the current study, whereas human umbilical vein endothelial cells were used to investigate eNOS phosphorylation. Second, fatty acids in the lipid emulsion (Intralipid) used for the treatment of drug toxicity are triglyceride forms, whereas free fatty acids were used in the current study. 9 Third, Intralipid contains several long-chain fatty acids, but we examined the effect of essential fatty acids on acetylcholine-induced relaxation. 9
In conclusion, linolenic acid attenuated acetylcholine-induced NO-mediated relaxation via the inhibition of NO-induced guanylate cyclase activation after NO formation. In addition, linolenic acid seems to contribute to the attenuation of severe vasodilation induced by a toxic concentration (3 × 10−4 M) of bupivacaine via the inhibition of the endothelial NO–cGMP pathway.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07043914 and NRF-2016R1D1A1B03930451).