
Abstract
Background:
Radiation therapy induces acute and chronic radiological toxicity, in particular hematological toxicity (HT). This study aimed to explore the mechanistic clue and potential predictors at the messenger RNA (mRNA) level.
Materials and Methods:
Peripheral blood was collected from 3 patients with cervical cancer (CC), nasopharynx cancer (NC), and tongue cancer (TC) after the first 2 Gy fraction of radiotherapy (RT). High-throughput sequencing was used to assess mRNA profiles.
Results:
Eleven genes, such as ALAS2(5-aminolevulinate synthase), SLC4A1(solute carrier family 4 member 1), HBG2(hemoglobin subunit gamma 2), TNFAIP3 (TNF α-induced protein 3), PER1 (period circadian clock 1), CCDC136 (coiled-coil domain containing 136), C9orf84 (chromosome 9 open reading frame 84), IL1B (interleukin 1β), FOSB (FosB protooncogene), NR4A2 (nuclear receptor subfamily 4), PARP15 (polymerase family member 15), had overlapping expression changes in all 3 cancers of which 3 (ALAS2, FOSB, and HBG2) are suggested as potential predictors for the early diagnosis of HT after RT.
Conclusions:
ALAS2, FOSB, and HBG2 may be useful predictors of HT in patients after RT. Eleven overlapping expression mRNAs among 3 cancers might be potential predictors for early diagnosis of radiation toxicity in patients.
Introduction
Radiotherapy (RT) refers to the application of ionizing radiation (IR), including radioactive isotopes or external beam RT, to treat patients with cancer by destroying cancer cells. 1,2 Since X-rays were first discovered in 1895, radiation has been widely used for cancer therapy; today, over 40% of patients with cancer undergo RT. 3,4 However, in recent years, many concerns regarding acute and chronic radiological toxicity, in particular hematological toxicity (HT), have emerged. Narendra et al reported that treatment interruptions among patients with medulloblastoma receiving craniospinal irradiation happened due to acute HT of RT. 5 A study by Olgun et al indicated the potential of intensity-modulated radiation therapy to induce long-term HT. 6 Bergsma et al. investigated persistent hematologic dysfunction (PHD) after radionuclide therapy among 274 patients with gastroenteropancreatic neuroendocrine tumors, and found that 2.9% of them developed secondary hematopoietic tumors, while 1.1% developed bone marrow failure. 7 Although a series of reports indicated no obvious HT to be induced by RT, the toxicities on other normal tissues have been observed. In a study reported by Hirotake et al, chemoradiotherapy, which is most widely used in the management of esophageal cancer, was found to cause severe acute toxicity in 30% to 50% of patients. 8 Ishikura et al also reported that using chemoradiotherapy to treat squamous cell cancer resulted in long-term toxicity, including pericarditis, heart failure, and radiation pneumonitis. 9 Most notably, epidemiological analysis suggests an increased HT risk after the start of palliative RT; 25% patients develop at least grade 2 HT. 10
Radiation and nuclear technology are being increasingly used in the fields of medicine, industry, and energy production, and the general public also face the potential risk of radiological exposure due to nuclear accidents, radiological terrorism, and even military conflicts. Therefore, in addition to the consideration of harmful side effects of radiotherapy to normal tissue, evaluation of individuals’ HT in response to various levels of radiological exposure is also valuable for identifying biological markers to identify the potential exposure individuals as well as enable early diagnosis of HT or clarification of radiation-exposed people and appropriate interventions. 11 Port et al used blood cell counts as a biomarker for clinical radiation injury and suggested that these counts could be used as a rapid predictor of such injury in the first 3 days after radiation exposure. 12 Ye et al evaluated the prognostic value of the following predictors in patients with nasopharyngeal carcinoma treated with RT: absolute neutrophil count (ANC), absolute platelet count, absolute lymphocyte count (ALC), neutrophil-to-lymphocyte ratio (NLR), and platelet-to-lymphocyte ratio (PLR). This analysis indicated that NLR and PLR were useful and cost-effective predictors that could be applied to inform more precise treatment strategies. 13 However, although a number of predictors have been identified in the past few decades, few epigenetic predictors have been developed.
Recently, messenger RNA (mRNA) expression profiling in cells or tissues has emerged as a potential means of discovering new predictors associated with RT and radiological toxicity. Nagaraja et al found that mRNA expression levels for E-cadherin, transforming growth factor β, and Snail were altered after radiation, potentially regulating radiation-induced epithelial–mesenchymal transition in lung cancer cells. 14 Zhou et al created a radiogenomic map linking computed tomographic image characteristics and mRNA expression profiles among patients with non-small-cell lung cancer and identified a number of highly coexpressed mRNAs associated with cancer prognosis and invasiveness. 15 Port et al measured changes in mRNA expression in baboon peripheral blood after irradiation ranging from 2.5 to 5 Gy using whole-genome microarrays. They found that 2128 mRNAs were upregulated after radiation, while 789 were downregulated of which 11 were verified by quantitative reverse transcription (qRT) polymerase chain reaction (PCR) to be significantly altered in baboons with hematological acute radiation syndrome. 16 Port et al also identified 29 radiation-inducible mRNAs from baboon peripheral blood samples, including CD177 (CD177 molecule), RNASE3 (ribonuclease A family member 3), and DAGLA (diacylgycerol lipase, alpha) diacylglycerol lip, which could be used as predictors to predict clinical RT response in humans. 17 Kim et al found 2 distinct mRNA expression profiles associated with different clinical behaviors in BRAF (B-Raf protooncogene, serine/threonine kinase) wild-type papillary thyroid carcinoma. 18 In short, mRNA expression profiling plays a critical role in understanding post-RT HT and identifying potential predictors. 19 -23 However, previous studies have focused mainly on toxic effects in cell line or animal models; few have conducted mRNA expression profiling using the peripheral blood cells directly from patients with cancer collected before and after RT.
In the present study, we hypothesized that mRNA sequence profiles might differ before versus after RT. We used high-throughput sequencing technology combined with bioinformatics tools to identify and compare mRNA expression profiles among 3 types of cancers: cervical cancer (CC), nasopharynx cancer (NC) and tongue cancer (TC). High-throughput sequencing, a powerful tool for characterizing transcriptomes, has attracted increasing attention as a means of identifying the mRNAs involved in the development of various cancers. 24 We selected these 3 cancer types for 2 main reasons: First, they are mainly treated by RT, and second, by comparing mRNA expression among cancer types, we hoped to identify common predictors among different type of cancers that might provide insight into the mechanisms of HT to enable early clinical interventions.
Materials and Methods
Baseline Patient Information
Three participants recently diagnosed with CC, NC, and TC were recruited into this study at the First Affiliated Hospital of the University of South China. The baseline information of each enrolled patient was as follows: Patient #1 was diagnosed with stage III-B differentiated squamous cell CC on April 4, 2017, and was a 55-year-old female with a body weight of 50 kg. Patient #2 was diagnosed with T4N2M0 stage IV-A undifferentiated nonkeratosis of NC on April 4, 2017, and was a 63-year-old male with a body weight of 62.5 kg. Patient #3 was diagnosed with poorly differentiated squamous cell TC (pT1N2bM0, stage IV-A) on April 4, 2017, and was a 53-year-old male with a body weight of 50 kg. All enrolled patients were confirmed to have no complications (Supplementary Table 1). All diagnoses were based on case histories, clinical symptoms, predictors, and/or tissue biopsy according to the National Diagnosis Criteria. All 3 patients received 2 Gy first fraction RT after diagnosis, and peripheral blood (at least 5 mL) was collected within 16 hours before and after the first fraction of RT. PAXgene blood RNA tubes were purchased from BD Biosciences (San Jose, California) and used to store blood samples at –70°C until use for high-throughput sequencing analysis. After samples were collected, we sent them to RiboBio Co, Ltd (Guangzhou, China) for immediate high-throughput sequencing analysis.
The study was performed in accordance with the Declaration of Helsinki and was approved by the First Affiliated Hospital of the University of South China (IRB approval no. NHUH-2017-28). All enrolled patients signed informed consent forms.
Total RNA Extraction and Complementary DNA Synthesis
Peripheral blood samples were collected before and after 2 Gy of the first fraction RT and immediately into the RNA extract reagent (QIAamp RNA blood Mini Kit; QIAGEN, Hilden, Germany), total RNA was extracted following the manufacturer’s instructions. After RNA extraction, concentrations were determined using the NanoDrop-2000 spectrophotometer (Thermo Scientific, Waltham, Massachusetts). The purity of extracted RNA was determined using the RNeasy Mini Kit (QIAGEN). Complementary DNA (cDNA) libraries were established using the SMARTer PCR cDNA synthesis kit with at least 500 ng of total RNA by 5’ rapid amplification of cDNA ends as described previously. 25
T-cell Receptor Sequencing (TCR) Library Establishment and Sequencing Analysis
Briefly, 2 rounds of PCR amplification were conducted. The first round was performed by the following process: Advantage 2 Polymerase mix (Clontech, Fremont, California) was used to synthesize cDNA. The nested universal primers (synthesized by Clontech) were 5’-AAGCAGTGGTATCAACGCAGAGT-3’ as well as the T-cell Receptor Sequencing (TCR) β constant region-specific primer 5’-AGATCTCTGCTTCTGATGGCT-3’. The PCR amplification conditions were as follows: 35 cycles of denaturing at 94°C for 15 seconds, followed by 3 minutes at 94°C, annealing at 58°C for 30 seconds, extension at 72°C for 45 seconds with a final extension at 72°C for 10 minutes. Then, 2% polyacrylamide gel was used for electrophoresis, and purification was carried out using the Gel Extraction Kit. Then, 2 μg cDNA from each sample was used to create the TCR sequencing library. Libraries were then sequenced on the HiSeq 2500 platform (Illumina, San Diego, California). Briefly, the analysis process was as follows: Raw data were stored in fastq format; after data filtering and quality control, the IMGT (ImMunoGeneTics) database was used for read mapping; genes were translated to amino acid sequences, and alignment was performed using the IMGT database. Finally, statistical analysis was performed on the results of the alignment.
Gene Ontology Analysis and Kyoto Encyclopedia of Genes and Genome Pathway Analysis
To explore the ontology of differentially expressed mRNAs/genes among the 3 types of cancers, we conducted Gene Ontology (GO) analysis. First, we conducted GO analysis on differentially expressed mRNAs and then on coexpressed mRNAs. The GO results were categorized as “biological processes,” “cellular components,” and “molecular functions.”
Kyoto Encyclopedia of Genes and Genomes (KEGG) is an integrated bioinformatics database that can be used to predict the pathways associated with genes and molecules. We conducted pathway enrichment analysis for differentially expressed mRNAs and mRNAs with overlapping expression among the 3 types of cancer.
Statistical Analysis
The fold change in mRNA expression levels before and after RT is presented in log2 format. P values and q values were used to analyze differences before and after RT. Pearson correlation coefficients were used to analyze the linear correlation between pairs of samples. P values <.05 were considered statistically significant. R software (R Development Core Team, Vienna, Austria) was used for differential expression and coexpression analysis as well as for GO and KEGG analysis.
Results
Differentially Expressed mRNAs/Genes Before and After RT in 3 Patients With CC, NC, and TC
In samples collected from the patient with CC before and after 2 Gy RT, we identified 35 771 mRNAs of which 621 were differentially expressed at a significance threshold of P < .01. In the patient with NC, 35 428 were identified of which 593 mRNAs were differentially expressed at a significance threshold of P < .01. In the patient with TC, we identified 36 575 mRNAs of which 395 were differentially expressed at a significance threshold of P < .01. Table 1 shows the top 20 upregulated mRNAs in peripheral blood cells of these patients with 3 types of cancer after the first 2 Gy fraction of RT. Tachykinin 4 (TAC4, NM_001077504.1) exhibited the highest fold change in CC; immunoglobulin super family member 9 (IGSF9, NM_020789.3) had the highest fold change in NC; and structural maintenance of chromosomes 1B (SMC1B, NM_148674.4) had the highest fold change in TC (P < .05). Table 2 shows the top 20 downregulated mRNAs after the first 2 Gy fraction of RT. Inhibitor of DNA binding 1 (ID1, NM_002165.3) had the greatest fold change in expression in CC (-6.523); C-C motif chemokine ligand 3 like 3 (CCL3L3, NM_001001437.3) had the greatest fold change in expression in NC (-4.507); and sulfotransferase family 1C member 4 (SULT1C4, NM_006588.3) had the greatest fold change in expression in TC (-3.889).
Top 20 Upregulated Genes / mRNAs in the Peripheral Blood Cells of Patients With 3 Types of Cancer After First 2 Gy Fraction of Radiotherapy.

Abbreviation: mRNA, messenger RNA.
Top 20 Downregulated Genes / mRNAs in the Peripheral Blood Cells of Patients With 3 Types of Cancer After the First 2 Gy Fraction of Radiotherapy.
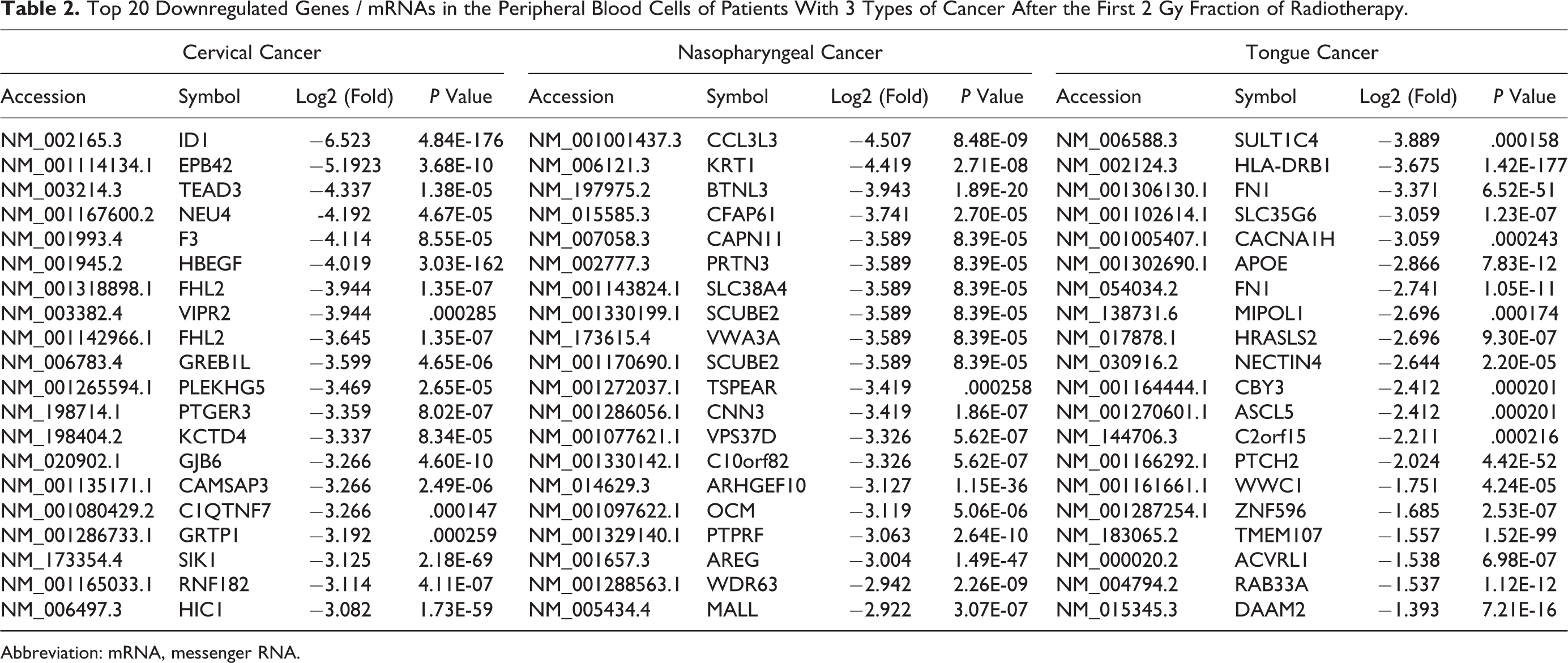
Abbreviation: mRNA, messenger RNA.
Coexpressed mRNAs/Genes Before and After RT Among Patients With 3 Types of Cancer
We determined the overlapping differentially expressed mRNAs in pair wise combinations of cancers: CC and NC, CC and TC, and NC and TC. A Venn diagram was constructed using R software to visualize the coexpressed mRNAs (Figure 1A). It was found that 51 overlapping expression changed mRNAs were identified in both CC and TC, 76 mRNAs had overlapping expression change in NC and TC, and 136 mRNAs had overlapping expression change in NC and CC. In addition, 445differentially expressed mRNAs were found specifically in patients with CC, 302 in patients with NC and 279 in patients with TC. Most importantly,11 mRNAs including ALAS2, SLC4A1, HBG2, TNFAIP3, PER1, CCDC136, C9orf84, IL1B, FOSB, NR4A2, PARP15 had overlapping expression among all the 3 cancer types after RT. (Table 3). In these 11 mRNAs, we found 3 co-upregulated genes c9orf84, IL1B, and FOSB and 6 co-downregulated genes ALAS2, SLC4A1, HBG2, TNFAIP3, PER1, and CCDC136. However, NR4A2 and PARP15 were found upregulated in patients with both NC and TC but downregulated in case with CC.

Differentially expressed messenger RNAs (mRNAs) in the peripheral blood cells of 3 patients with different type of cancers after the first 2 Gy fraction of radiotherapy. A, Venn diagram showing distinct and overlapping differentially regulated mRNAs among three types of cancers. B, Gene ontology (GO) analysis of 11 mRNAs differentially expressed in all three types of cancers.
The Overlapped Genes Differentially Expressed Among Patients With 3 Types of Cancer After the First 2 Gy Fraction of Radiotherapy.a

alog2(FC) = log2(Fold of change).
Gene Ontology Analysis
The GO analysis results for the 3 cancer types are shown in Figure 2. In TC, differentially expressed mRNAs were predicted to have the following main functions: cell adhesion, biological adhesion, cell–cell adhesion, hemoglobin complex, extracellular matrix part, basement membrane, and cell soma in the biological process category; cell periphery and plasma membrane in the cellular component category; and cell–cell adhesion, cell adhesion, and biological adhesion in the molecular function category (Figures 2 –4). In NC, differentially expressed mRNAs were predicted to perform molecular functions related to regulation of RNA metabolic process, positive regulation of cellular biosynthetic process, positive regulation of macromolecule biosynthetic process in the biological process category; nuclear lumen, intracellular organelle lumen in the cellular component category; and DNA binding, transcription regulator activity, and zinc ion binding in the molecular function category (Figure 2). In CC, some mRNAs played roles in transcription factor activity, sequence-specific DNA binding and nucleic acid binding, and transcription factor activity. Some other mRNAs had putative functions related to the response to lipids and steroid hormones (Figure 3). In TC, mRNAs played roles in cell adhesion, biological adhesion, cell motion, hemoglobin complexes, cell soma calcium ion binding, and cation binding (Figure 4).We conducted GO analysis on these 11 genes of overlapping expression changes among patients with all 3 cancers and found that 4 genes were related to the positive regulation of transcription from RNA polymerase Ⅱ promoter. Three genes were predicted to play a role in the response to cyclic adenosine monophosphate, 2 were related to cellular components, and 2 were predicted to play a role in kinase binding (Figure 1B).

Gene ontology (GO) analysis of the differentially expressed messenger RNAs (mRNAs) found in tongue cancer (TC).

Gene ontology (GO) analysis of the differentially expressed messenger RNAs (mRNAs) found in cervical cancer (CC).

Gene ontology (GO) analysis of the differentially expressed messenger RNAs (mRNAs) found in nasopharynx cancer (NC).
Kyoto Encyclopedia of Genes and Genomes Analysis
The results of KEGG analysis are shown in Figure 5. In CC, 16 mRNAs were involved in the herpes simplex infection pathway, 14 in the influenza A pathway, and 16 in the cytokine–cytokine receptor interaction pathway (Figure 5A). In NC, 140 mRNAs were involved in the viral carcinoma pathway, 123 in the herpes simplex infection pathway, and 113 mRNAs in the spliceosome pathway (Figure 5B). In TC, 13 mRNAs were involved in the human T-lymphotropic virus type I infection pathway, 11 in the regulation of actin cytoskeleton, and 10 in the Wnt signaling pathway (Figure 5C). Furthermore, we conducted KEGG analysis on these 11 genes of overlapping expression changes among patients with 3 cancers. We identified 3 pathways associated with these mRNAs: the TNF signaling pathway, nucleotide binding oligomerization domain (NOD)-like receptor signaling pathway, and nuclear factor κB signaling pathway (Supplemental Figures 1–3).
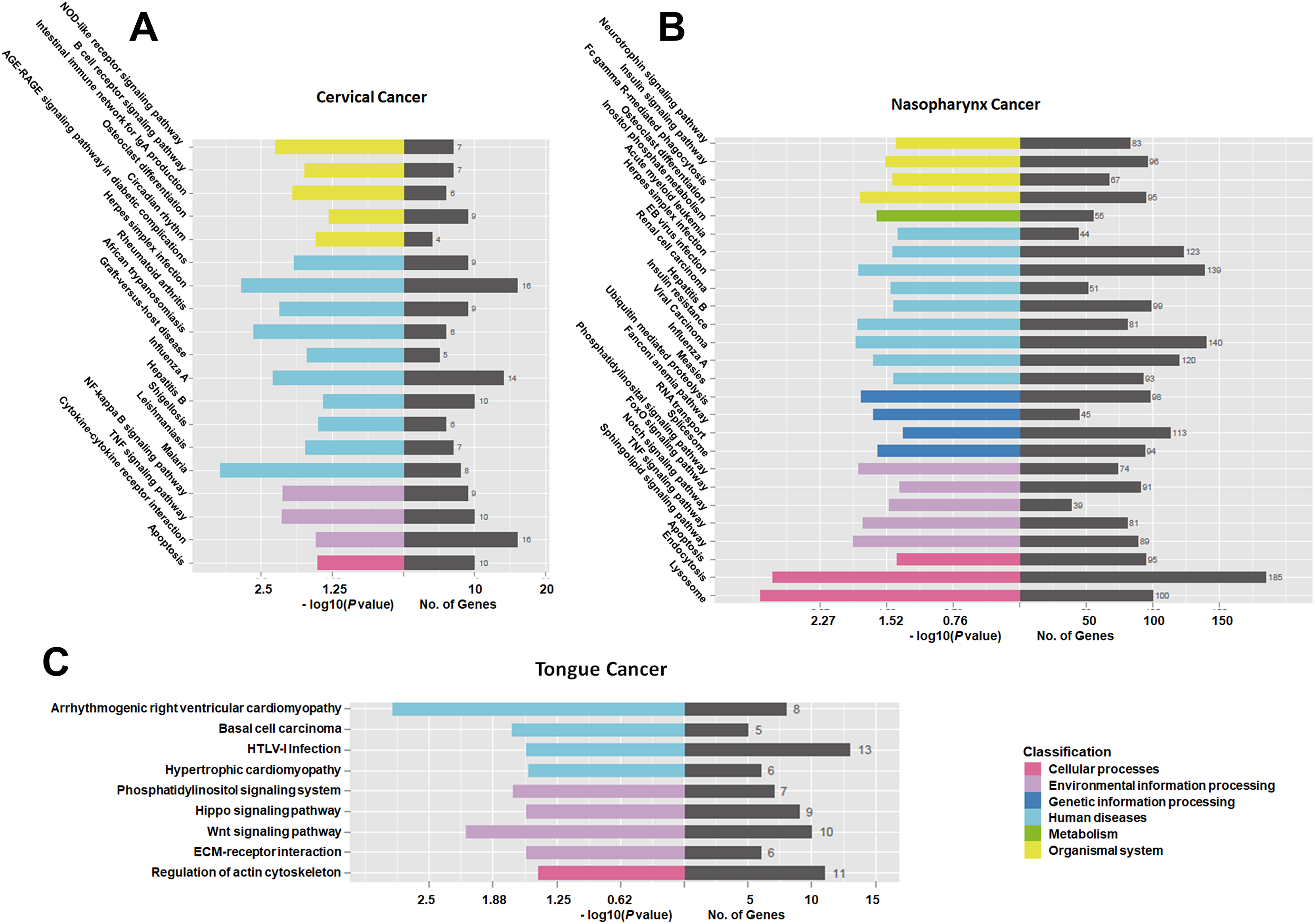
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of the differentially expressed messenger RNAs (mRNAs) found in tongue cancer (TC), cervical cancer (CC), and nasopharynx cancer (NC).
Discussion
General Finding
The importance of mRNAs as clinical predictors has long been recognized. Many studies have investigated the alteration or deregulation of various genes that can fundamentally affect the processing of the molecular transcriptome and proteome. 26,27 However, most research on cancer-associated mRNAs has focused mainly on single mRNAs or signaling pathways; few studies have focused on mRNA expression in patients with cancer before and after RT. To explore the effects of RT on mRNA expression, we performed high-throughput sequencing analysis on samples from 3 common clinical cancers. We found that, after RT, 445 mRNAs were significantly differentially expressed only in patients with CC, 302 in patients with NC, and 279 in patients with TC. Importantly, we have identified 11 genes such as ALAS2, SLC4A1, HBG2, TNFAIP3, PER1, CCDC136, C9orf84, IL1B, FOSB, NR4A2, and PARP15 that had overlapping expression changes in all 3 cancers of which 3 (ALAS2, FOSB, and HBG2) are proposed as predictors of HT associated with RT.
TAC4, IGSF9, and SMC1B were the Most Upregulated mRNAs in CC, NC, and TC
The most upregulated mRNAs in CC, NC, and TC were TAC4, IGSF9, and SMC1B, respectively. It has been reported that human hemokinin-1 (hHK-1), and its truncated form hHK-1(4-11), are encoded by TAC4. These proteins have inhibitory effects on cellular proliferation in the human promyelocyte leukemia cell line HL-60 and have been considered as immunomodulatory factors in cancer chemotherapy. 28 IGSF9 was cloned and characterized in 2002 and is a member of the immunoglobulin superfamily expressed in the developing nervous system (in turn reported to have various functions in nerve tissues). 29,30 Ansari et al used high-definition mass spectrometry to show that SMC1B (structural maintenance of chromosomes 1B) was linked with the development of pancreatic cancer: It was significantly upregulated in serum samples from patients with pancreatic cancer. 31
4.3 SULTIC4, IDL, and CCL3L3 were the Most Downregulated mRNAs in CC, NC, and TC
The most downregulated mRNAs in CC, NC, and TC were ID1 (inhibitor of DNA binding 1), CCL3L3 (C-C motif chemokine ligand 3 like 3), and SULT1C4(sulfotransferase family 1C member 4), respectively. ID1 has been reported to be a biomarker for self-renewing tumor cells in glioblastoma, and its expression is associated with resistance to radiation-induced DNA damage. 32 Downregulation of ID1 may lead to an increasing sensitivity of blood cells to subsequent irradiation of RT and thus potentiate the radiotoxicity. CCL3L3, a member of the human CC chemokine family, is a mediator exhibiting a variety of proinflammatory activities, such as chemotaxis and the activation or proliferation of lymphocytes and macrophages. Altered CCL3L3 expression is linked with tumorigenesis and the progression of cancer. 33 However, it has not previously been reported that CCL3L3 is downregulated after RT. SULT1C4, a member of the cytosolic sulfotransferase family, has been shown to sulfonate critical compounds, such as estrogens. 34
Eleven mRNAs Commonly Altered Among All 3 Cases
When we attempted to figure out the overlapping expressed mRNAs either in both paired patients with cancer or in all the three patients with cancer, we found that several differentially expressed genes/mRNAs were expressed in more than one type of cancer. Among pairs of cancer types, differentially expressed genes were varied greatly. For instance, C9orf84, CREM, and FOSB were differentially expressed in CC and TC; AGRN, AKAP6, and ALAS2 were differentially expressed in CC and NC; and A2M, ABR, and AFF2 were differentially expressed in TC and NC. Furthermore, some mRNAs were upregulated in one cancer type but downregulated in another or vice versa. For instance, AGRN was upregulated in CC but downregulated in NC, while ACVR1C was upregulated in CC but downregulated in TC. One explanation for these results may be that genes may play different biological roles in different cancers, another may be in the development of cancer biological process mediated by RT, and a set of molecular or factors may be involved into triggering different genes expression. The underlying molecular mechanisms associated with these functions and changes should be further elucidated. The present study also figured out 11 overlapping expressed genes among the patients with 3 types of cancer after the first 2 Gy fraction of radiotherapy including ALAS2, SLC4A1, HBG2, TNFAIP3, PER1, CCDC136, C9orf84, IL1B, FOSB, NR4A2, and PARP15. Among these 11 mRNAs, most of them are associated with cancer development. For instance, CCDC136 and IL1 may influence tumor development. TNFAIP3 is reported to be linked with the risk of autoimmune disease and acts as a “master switch” in the expression of inflammation factors 35 in the cancer development. PER1, a circadian rhythm-related gene, has been reported to be a tumor suppressor in human oral squamous cell carcinoma. 36
ALAS2, FOSB, and HBG Were Proposed to be the Potential Predictors of HT Associated With RT
To compare the predictors for radiation exposure, Tichy et al collected peripheral blood samples from patients with endometrial or head and neck cancer undergoing radiation and assessed gene expression patterns using multiplex qRT-PCR analysis. The authors identified 5 transcriptional predictors whose expression levels changed significantly after radiation exposure (P < .05). 11 Wen et al characterized gene expression patterns in peripheral lymphocytes among patients with acute lymphoid leukemia after 2 RT sessions with 4.5 Gy radiation and found that 478 genes were significantly differentially expressed. 37 In the present study, for some of these genes (eg, CCL3L3), which is the first to identify RT-related changes in expression in peripheral blood, the expression changes in mRNA level might be specifically mediated by radiation, which is suggested to be potential predictors for radiation exposure. Notably, we found that several differentially expressed genes were associated with HT, including adult acute lymphoblastic leukemia. For instance, ferrodoxin reductase (FDXR), which modulates several components of the iron pathway, is significantly upregulated in patient blood samples after radiation exposure. 38 The differentially expressed genes associated with hematological defects in the present study should be researched further to determine the mechanisms underlying these associations. However, the patterns of gene expression identified in the present study differed from those of previous studies. This may be due to disparities in cancer types or racial characteristics of the patients or radiation dosage. Most importantly, 3 overlapping expressed genes (ALAS2, FOSB, and HBG2) are suggested as potential predictors for the early diagnosis of HT after RT based on the considerations as follows. It was reported that ALAS2 overexpression could rescue an alteration in the heme biosynthesis transcriptional profile that was repressed by transcriptional regulator Bach1. 39 ALAS2 mutations can cause X-linked sideroblastic anemia, leading to defective heme synthesis and ineffective erythropoiesis. 40 FOSB was recently shown to play a role in oncogenic activation, especially in endothelial proliferation. 41 HBG2 polymorphisms are associated with sickle cell anemia and inherited bone marrow failure syndromes. 42,43 However, these genes practically associated with the HT should be analyzed by further cell or epidemiological studies to uncover underlying mechanisms.
Gene Ontology and KEGG Analysis Predicted Biological Functions of 11 Overlapping Genes
For those 11 overlapping expressed mRNAs in all the 3 cancer types, we also conducted GO and KEGG analyses to gain insight into the biological functions of these 11 genes of common expression changes. Our results showed that these genes were all associated with the terms “biological process,” “molecular function,” “cellular component,” and “signaling pathways,” and all were associated with tumor development and poor prognosis. Specifically, the biological functions such as blood microparticle, either positive or negative regulation of transcription from RNA polymerase Ⅱ promoters, and hemoglobin complex should be drawn attention since these predicted functions are involved in the HT mediated by RT. Signaling pathways including TNF signaling pathway, NOD-like receptor signaling pathway, and NF-κB signaling pathway for these 11 overlapping genes are also very important to direct the next step study design. The fact that these 11 genes were expressed in all 3 cancer types suggests that different types of cancer may be regulated by similar genes. We suggest that the mRNA levels for these genes (in particular those with hematological effects, including ALAS2, FOSB, and HBG2) may be useful as predictors for the early diagnosis of HT after RT. Considering that these 3 patients performed partial body irradiation, and different radiation areas may affect the results of mRNA expression profile, 44 an in-depth characterization of these genes in more collected samples should be required in order to confirm the possibility to be as predictors of HT after RT. Another attention that should be mentioned is targeted effects and nontargeted effects originated from the irradiated tumors cells when the IR performs action on the blood cells. 22 Further studies should be designed to investigate the role of these mRNAs in this bystander effects. Moreover, according to the KEGG analysis, a number of virus infection-related pathways were potentially associated with radiation response in patient with CC. This maybe the virus infection-related pathways are similar to the inflammation reaction pathway, which has been identified after IR exposure. However, the underlying mechanism regarding this needs to be investigated further.
Limitations
It is important to mention several limitations of the present study. First, we collected only a limited number of samples and did not analyze an additional set of samples using qRT-PCR to validate our results. Second, we recruited only 1 patient per cancer type. Finally, it is possible that mRNA levels may not correspond perfectly to protein abundance; further studies should clarify the relationship between mRNA levels and protein levels. Although our study was performed on only 3 patients and our results were not validated by qRT-PCR, it provided some clinical information that mRNA predictors may be useful for the early detection of radiation-induced HT or other types of radiation-induced cytotoxicity.
Conclusion
The mRNA expression profile in peripheral blood cells from 3 patients with cancer was compared between prior and post 2 Gy irradiation using high-throughput sequencing. A large number of differentially expressed genes/mRNAs were identified of which 11 genes including ALAS2, SLC4A1, HBG2, TNFAIP3, PER1, CCDC136, C9orf84, IL1B, FOSB, NR4A2, and PARP15 were commonly altered in all 3 cancers. Furthermore, through GO and KEGG analysis, ALAS2, FOSB, and HBG were proposed to be as the potential predictors of HT associated with RT. However, further research is required to expand the sample size to validate and elucidate the molecular function of these mRNAs and the mechanisms underlying the effects of RT on mRNA expression.
Supplemental Material
Supplementary-table1 - Analysis of mRNA Expression Patterns in Peripheral Blood Cells of 3 Patients With Cancer After the First Fraction of 2 Gy Irradiation: An Integrated Case Report and Systematic Review
Supplementary-table1 for Analysis of mRNA Expression Patterns in Peripheral Blood Cells of 3 Patients With Cancer After the First Fraction of 2 Gy Irradiation: An Integrated Case Report and Systematic Review by Yue-Hua Nie, Xiao-Dan Liu, Ruixue Huang, Da-Fei Xie, Wen-Jun Yin, Hua Guan, Zi-Jian Yu, and Ping-Kun Zhou in Dose-Response
Supplemental Material
Supplement_Figure_1 - Analysis of mRNA Expression Patterns in Peripheral Blood Cells of 3 Patients With Cancer After the First Fraction of 2 Gy Irradiation: An Integrated Case Report and Systematic Review
Supplement_Figure_1 for Analysis of mRNA Expression Patterns in Peripheral Blood Cells of 3 Patients With Cancer After the First Fraction of 2 Gy Irradiation: An Integrated Case Report and Systematic Review by Yue-Hua Nie, Xiao-Dan Liu, Ruixue Huang, Da-Fei Xie, Wen-Jun Yin, Hua Guan, Zi-Jian Yu, and Ping-Kun Zhou in Dose-Response
Supplemental Material
Supplement_Figure_2 - Analysis of mRNA Expression Patterns in Peripheral Blood Cells of 3 Patients With Cancer After the First Fraction of 2 Gy Irradiation: An Integrated Case Report and Systematic Review
Supplement_Figure_2 for Analysis of mRNA Expression Patterns in Peripheral Blood Cells of 3 Patients With Cancer After the First Fraction of 2 Gy Irradiation: An Integrated Case Report and Systematic Review by Yue-Hua Nie, Xiao-Dan Liu, Ruixue Huang, Da-Fei Xie, Wen-Jun Yin, Hua Guan, Zi-Jian Yu, and Ping-Kun Zhou in Dose-Response
Supplemental Material
Supplement_Figure_3 - Analysis of mRNA Expression Patterns in Peripheral Blood Cells of 3 Patients With Cancer After the First Fraction of 2 Gy Irradiation: An Integrated Case Report and Systematic Review
Supplement_Figure_3 for Analysis of mRNA Expression Patterns in Peripheral Blood Cells of 3 Patients With Cancer After the First Fraction of 2 Gy Irradiation: An Integrated Case Report and Systematic Review by Yue-Hua Nie, Xiao-Dan Liu, Ruixue Huang, Da-Fei Xie, Wen-Jun Yin, Hua Guan, Zi-Jian Yu, and Ping-Kun Zhou in Dose-Response
Footnotes
Authors’ Note
PKZ contributed to study concept and critical design; YHN, ZJY, XDL, DFX, WJY, ZJY, HG conducted the experiments; YHN, ZJY, XDL, RXH, DFX, WJY, ZJY acquired, analyzed, and interpreted data; YHN and PKZ fulfill the initial manuscript; and PKZ critically reviewed and revised the final manuscript. All authors were involved in the current study, contributed to this study.
This study was performed in accordance with the Declaration of Helsinki and was approved by the First Affiliated Hospital of the University of South China (IRB approval no. NHUH-2017-28). All enrolled patients signed informed consent forms.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is supported by grants from National Key Basic Research Program (973 Program) of MOST, China (Grant No. 2015CB910601), the National Natural Science Foundations of China (Grant No. 31530085, 31870847, U1432248, U1803124), and Natural Science Foundation of Hunan Province (Grant No. 2018JJ3166)
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.