
Abstract
Brain development is an organized, but constantly adaptive, process in which genetic and epigenetic signals allow neurons to differentiate, to migrate, and to develop correct connections. Gender specific prenatal sex hormone milieu participates in the dimorphic development of many neuronal networks. Environmental cues may interfere with these developmental programs, producing adverse outcomes. Bisphenol A (BPA), an estrogenic/antiandrogenic endocrine disruptor widely diffused in the environment, produces adverse effects at levels below the acceptable daily intake. This review analyzes the recent literature on the consequences of perinatal exposure to BPA environmental doses on the development of a dimorphic brain. The BPA interference with the development and function of the neuroendocrine hypothalamus and of the nuclei controlling energy balance, and with the hippocampal memory processing is also discussed. The detrimental action of BPA appears complex, involving different hormonal and epigenetic pathways activated, often in a dimorphic way, within clearcut susceptibility windows. To date, discrepancies in experimental approaches and in related outcomes make unfeasible to translate the available information into clear dose–response models for human risk assessment. Evaluation of BPA brain levels in relation to the appearance of adverse effects in future basic studies will certainly give better definition of the warning threshold for human health.
Keywords
Introduction
At the beginning of this review, it is essential to underline that even though the tolerable daily intake of 50 μg/kg body weight is still thought to be safe and considered the reference dose for in vivo studies (Shelnutt et al. 2013), it is now clear that the dose response effect for bisphenol A (BPA) is not monotonic, and that some disruptive effects may also be seen at lower dosages (see other articles of this special issue). Moreover, even at low doses, the risk of negative outcomes is much higher during development than in adulthood because of the greater exposure of the fetus. Among the possible causes of this high exposure are the efficient placental transfer of both BPA and its glucuronide derivative, coupled to the deficiency of fetal UDP glucuronosyltransferase (UGT) enzyme (Nishikawa et al. 2010). The combination of these factors brings to a consistent deconjugation of BPA-glucuronide, devoid of estrogenic activity, to active estrogenic BPA in the fetal compartment.
The majority of the studies on the developmental origin of adult adverse outcomes use laboratory animals (rats and mice). With this approach, many important molecular mechanisms of BPA action have been clarified, and specific adverse effects at doses well below the environmental exposure have been discovered as well (Rubin 2011). Yet, because of a series of divergence, it is still not possible to translate all this information into a clear dose–response model for human risk assessment. Actually, wide differences exist in environmental daily exposure and route of administration (mainly from food consumption in humans, from venous, intraperitoneal, or oral—gavage or dietary pellets—administration in rodents), time and length of exposure (almost undetermined in humans, variable in rodents—from the beginning of gestation, only in the prenatal period, only for few days, from a given day of pregnancy to a given day of postnatal life, and so on), species-specific BPA pharmacokinetic, metabolism, and possibly differences in placental transfer.
The route of administration seems to be a fundamental parameter to be taken in consideration, since it has been demonstrated that BPA accumulation in the different fetal rat tissues as a function of a single maternal administration is similar in fetal and maternal compartments only if injected intravenously; oral administration of the same dose does not produce measurable levels of BPA in any of the fetal tissues (Doerge et al. 2011).
To better define the relationship between exposure levels and risk, and to determine a possible risk threshold, it is important to know the extent of BPA accumulation in the various tissues as a function of the negative consequences recorded. Unfortunately, almost none of the studies evaluating the impact of prenatal exposure on postnatal/adult outcomes assessed BPA accumulation in the exposed animals, neither the effect of gender on accumulation. These parameters would be particularly important in central nervous system (CNS) studies, as BPA preferentially accumulates in the fetal brain (Doerge et al. 2011; Mita et al. 2012), where its average concentration is directly related to the dosage used and it is higher in males than in females (Mita et al. 2012).
In this review, the basic mechanisms of CNS differentiation will be summarized, focusing on the formation of a dimorphic brain in rodents and humans and the influence of BPA on these processes. The impact of BPA prenatal exposure on the neuroendocrine control of the endocrine axes, on the central mechanisms regulating food intake and on higher functions, providing the possible molecular mechanism of action will then be discussed. Due to the large amount of articles dealing with BPA and brain, the discussion will be limited to the recent literature referring almost exclusively to studies in rodents and to the effects observed at exposure doses equal to or lower than the tolerable daily intake for humans (referred as “low BPA”). Finally, as sex dimorphism in brain functions is widely diffused in CNS physiology, possible gender differences in BPA action will also be reported, whenever available.
Prenatal Exposure to BPA and Brain Functions
Over the last years, there has been an increasing effort to better characterize the long-term impact of early exposure to BPA on brain organization. The research in this field, performed mainly in rodents, identified several adverse outcomes on brain development, differentiation, and function, which have raised a growing concern that similar effects might occur also in humans. Indeed, BPA exposure is ubiquitous and continuous in humans, as the compound or its metabolites have been found at a mean concentration of 1 μg/L in more that 95% of human urine samples (Calafat et al. 2005).
Albeit BPA exposure affects CNS physiology at all life stages, the vulnerability is higher in the developing brain due to the lipophilic chemical structure that allows it to easily cross the blood–placental and blood–brain barriers (Nishikawa et al. 2010; Sun et al. 2002), as well as to be delivered to offspring through lactation (Zimmers et al. 2014). Moreover, as opposed to an adult exposure, the interference with the early phases of CNS embryonic/fetal/neonatal development makes permanent changes that prime and alter brain physiology not only in the exposed animals, but even in their progeny (Doerge et al. 2010; Manikkam et al. 2013). Furthermore, due to its estrogenic and antiandrogenic activities (Delfosse et al. 2014), BPA can interfere with the dimorphic development of the neuronal networks controlling many endocrine systems and brain functions (see below). As a matter of fact, sex differences have been well-documented not only in the mechanisms controlling reproduction, but also in nonreproductive behaviors (see Jazin and Cahill 2010 and Wolstenholme et al. 2011 for references).
It should be underlined that despite the abundance of literature, there is no general agreement on specific long-term effects of prenatal exposure on a given brain parameter and very often it is difficult to compare the results because of differences in the experimental designs. Nevertheless, at present, it is a shared opinion that BPA is one of the potential candidates for hazardous prenatal exposure.
Basic Mechanisms of Brain Development and Involvement of BPA on These Processes
The time scale in brain maturation is clearly very different between rodents and humans; yet, the sequence of the key events is largely stable, allowing a short description of the main hallmarks similar in the 2 species. Brain development during embryogenesis begins with the differentiation of neural progenitor cells (approximately at mid-gestation in rodents and in the third gestational week in humans) and is characterized by a series of dynamic and adaptive processes that promote the emergence and the differentiation of the various neuronal structures. These processes operate within a highly constrained and organized context, which, however, constantly changes following a continuous program of specification and refinement. The main event in this first phase is the proliferation of neuronal progenitors followed by the establishment and maturation of the 2 main brain cell lineages (neurons and glia). These cells then migrate from the proliferative ventricular zone to their final destinations, where neurons begin to produce neurotransmitters and neurotrophic factors as well as to form and extend dendritic and axonal processes. To allow neuronal migration, specialized glial cells form scaffolding processes along which cells can move. By the end of embryogenesis, the main components of the CNS are developed, and the primitive map of the final brain organization is defined. During the following fetal period, neurons, guided by the local environment, establish synaptic contacts with other neurons. It is now clear that the hallmark of brain differentiation is the high capacity of adaptation, which is the result of a continuous adjustment of neuronal connectivity (formation and elimination of synaptic contacts) and neuronal number (loss by apoptosis of neurons that do not uptake enough neurotrophic factors from target neurons at synapses). These 2 processes are essential to achieve and establish the complex network of the developing CNS. The brain also continues to evolve postnatally, increasing its size and undergoing structural changes in both gray and white matter compartments till adulthood. These structural changes correspond to the modifications in the functional organization and in the resulting behaviors. For a more comprehensive overview of brain development, see Stiles and Jernigan 2010.
Beyond the activation of homeobox genes, the process of neurogenesis and maintenance of neuronal differentiation are ensured by the silencing or the switch on/off of the transcription of specific genes. This fast gene expression profile modification is achieved via epigenetic changes both at specific DNA sequences and in the surrounding chromatin. Even though epigenetic modifications are the hallmark of the differentiation period, the emerging consensus is that the brain is particularly sensitive to these processes during the entire life span, when DNA silencing/activation and/or chromatin remodeling are involved in many neuronal processes and complex behaviors (Champagne 2012).
The epigenome comprises a set of chromatin regulators, the most studied of which are DNA methylation/demethylation in cytosine rich regions of gene promoters (CpG islands) and posttranslational histone modifications (mainly acetylation or methylation). The epigenetic modifications, driven by specific enzymes such as DNA methyltransfereases (DNMTs) and histone remodeling enzymes (Jarid, SIRT1 and others), dynamically interact with each other to define the correct transcriptomic profile. This consists in the expression of the appropriate set of genes in specific cell phenotypes at correct time points (Szyf 2009), and is a process particularly sensitive to environmental cues (Reik 2007).
From this short description of the basic principles of neural development, it is clear that the process involves an interplay between genetic/epigenetic and environmental factors during the entire life: the programmed activation of gene transcription provides proteins essential for brain development and maintenance, while the environment provides inputs that shape and influence the direction of the emerging neuronal network and its activity. The long-lasting brain plasticity ensures a high degree of adaptation to external inputs but also makes the structure highly vulnerable to negative external influences including the exposure to diet, to maternal behavior, and to xenocompounds, among which endocrine disruptors (Szyf 2009).
The effects of BPA on the developing brain have been studied in vitro in pluripotent neural progenitors and in primary neuronal or glial cultures or in brain tissues/cells of prenatally exposed rodents. The very few results from the literature in which the adverse effect on neuronal development has been investigated in pluripotent cells demonstrate that BPA is able to disrupt neurogenesis by influencing neuronal stem cell proliferation and viability (Kim et al. 2007), by modifying stem cell fate (neuronal/glial progenitors ratio; Okada et al. 2008) and by promoting neuronal progenitor differentiation into neurons (Kim et al. 2009) and the acquisition of the final neuronal phenotype (Yokosuka et al. 2008). The same disruption of neurogenesis and neuronal migration has been observed in some brain areas also in embryo mouse after maternal exposure (Itoh et al. 2012; Komada et al. 2012; Mathisen et al. 2013); these effects possibly arise from the upregulation of several early genes involved in cell migration and differentiation (Itoh et al. 2012). As correct neuronal positioning is a prerequisite for the formation of normal circuitry, the BPA-induced modifications of these phenomena might lead to abnormal connections among brain areas.
Several evidences from in vitro and in vivo models have clearly established that BPA induces changes in the epigenetic programming of different peripheral tissues and cell types that endure throughout lifetime (Dolinoy et al. 2007, see also Singh and Li 2012 for references). Even though the impact of low BPA on brain epigenome during development is still to be fully clarified, Yaoi et al. (2008) demonstrate, by a genome-wide analysis, that BPA exposure is associated with the changes in the methylation degree of several genes, the methylation status of which is developmentally stage dependent. As messenger RNA (mRNA) expression of some of these genes changes with the development as well as with the exposure to BPA, it is conceivable that the compound, modulating DNA methylation, may directly affect the transcriptome. In line with this hypothesis, it has been demonstrated in vitro that a short exposure of embryonic hypothalamic neurons to micromolar doses of BPA may affect the expression of DNMTs (Warita et al. 2013; Kundakovic et al. 2013). Also modifications of histone remodeling have been associated with neurodevelopment defects (Contestabile and Sintoni 2013), and it has been shown that BPA modifies histone mark both in vitro and in vivo (Singh and Li 2012). However, the direct effects of the compound of neuronal histone profile during embryogenesis have never been evaluated.
Development of Brain Dimorphism in Rodents and Humans and Interference of BPA With This Process
The majority of the studies on brain sex differentiation focused on brain regions involved in the control of the reproductive system and of sex behavior, which are definitely different in size and function between males and females. The 2 best known dimorphic brain structures are the sexual dimorphic nucleus of the medial preoptic hypothalamic area (SDN-POA) in rodents, which correspond to the interstitial nucleus of the anterior hypothalamus (INAH) in humans, and the anteroventral periventricular (AVPV) nucleus. The first one controls male sex behavior and is larger in males than in females; the second one is critical for the cyclic control of ovulation and is larger in females than in males. In the last years, exponentially growing evidence in animals and humans highlighted that areas important for cognition, memory, and emotions, for energy balancing traits, as well as for sensorimotor and reward systems are dimorphic in structure and functions (Gillies and McArthur 2010; Sacher et al. 2013); thus brain sex dimorphism appears to be the norm rather than an exception in CNS physiology.
It is widely accepted that the developing brain is bipotential at the beginning, being equally capable to differentiate into a male or a female phenotype. The determinant variable is the different hormonal milieu to which the brain is exposed during a “critical sensitive window” of perinatal development. During this period (which spans between mid gestational and postnatal day 10 in rodents and between gestational weeks 8 and 24 in humans), a complex series of “organizational” events induce permanent modifications of the cell number and morphology of the hypothalamic and other nuclei, generating specific neuronal networks which represent the morphofunctional basis of the sex-specific response of the adult brain to hormonal and environmental inputs (“activational” effects). According to this model (summarized in Figure 1), the chromosomal sex of the embryo (sex chromosomes XX or XY) determines the development of the ovaries or the testes, respectively. Once determined the gonadal sex, brain feminization occurs in the absence of gonadal hormones. On the contrary, in males, a prenatal peak of testosterone from the fetal testes drives the developing brain toward a male phenotype. The effect of testosterone within the brain is dual: on one hand, it organizes neurons to promote a male-typical mode of action (process of masculinization), and on the other, it suppresses the female-typical mode of action (process of defenimization; Arnold 2009; Negri-Cesi et al. 2008). The mechanism of testosterone action on masculinization and defeminization differs between rodents and humans. In rodents, the conversion of testosterone to estradiol, through the enzyme aromatase, and the activation of estrogen receptors (either ERα or ERβ) is mandatory; even if not as important, also the formation of dihydrotestosterone (DHT), through the two 5α-reductase enzymes (types 1 and 2), and the activation of androgen receptors (ARs), play a significant role in some aspects of brain masculinization. Thus, both androgens and estrogens are needed to fully differentiate the rodent male brain, while the lack of gonadal hormones is enough to drive the structure in a female direction (see Colciago et al. 2009; McCarthy and Arnold 2011 and Negri-Cesi et al. 2008 for references).
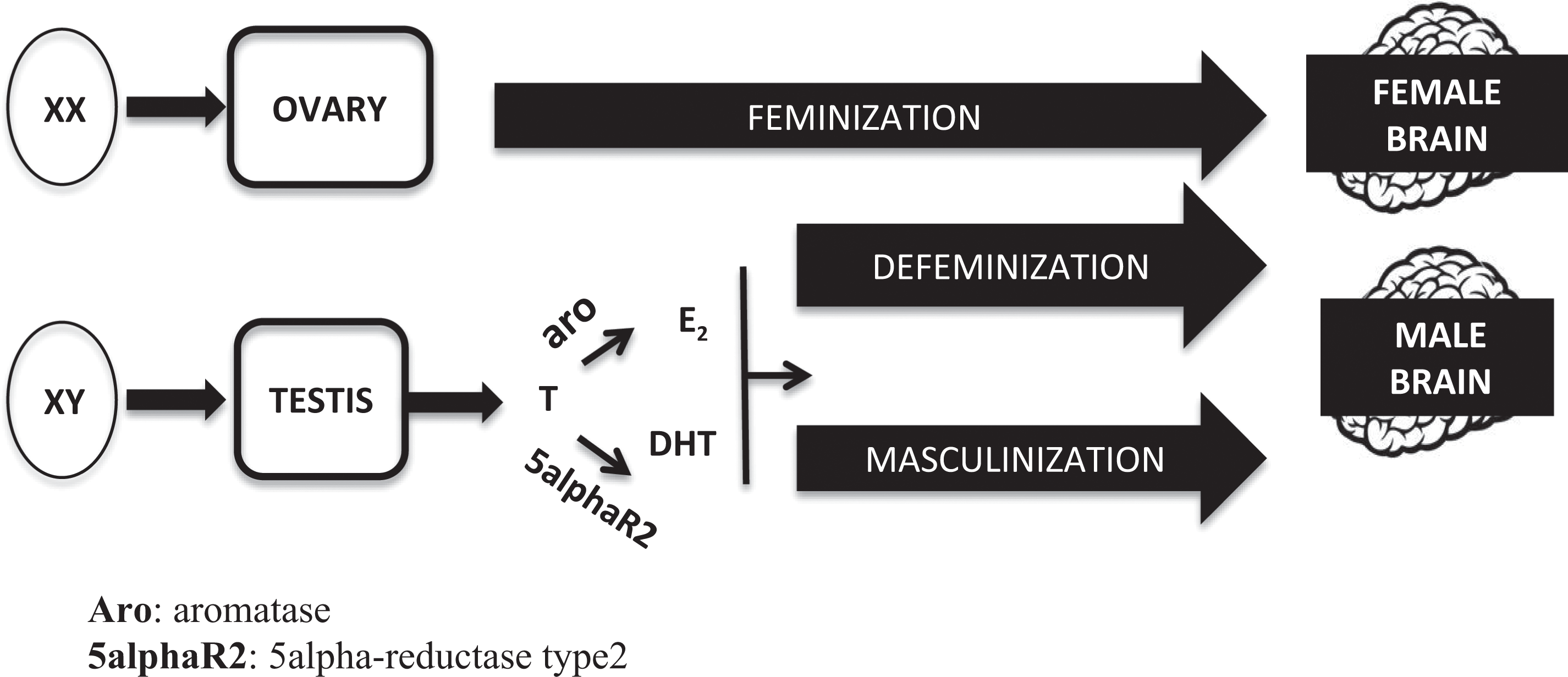
Classic model of the brain sexual differentiation process.
Whether the same mechanism has a role also in the human brain has not been clearly established yet. Some hypotheses inferred from specific phenotypes of human diseases that affect circulating testosterone or its mechanism of action suggest that estrogens do not have the same critical role as in rodents, and that the human fetal brain develops in a male direction thanks to a direct action of the testosterone-AR system on the developing nerve cells or in a female direction thanks to the absence of this hormone (Bao and Swaab 2011; Negri-Cesi et al. 2008).
In addition to prenatal hormones, some sex differences arise also from a direct genetic effect driven by genes located on X or Y chromosomes, as well as from sex specific effects of the environment triggered by epigenetic changes (McCarthy and Arnold 2011; Ngun et al. 2011). Interestingly, epigenetic modifications also mediate at least part of the mechanism of action of testosterone on brain sex differentiation. A picture of a tight interaction among genetic sex, sex steroid hormones, and the epigenome in shaping the dimorphic brain is now emerging. Some proofs of this interplay are (1) the methylation degree of sex steroid hormone receptor promoters is dimorphic (Westberry et al. 2010); (2) the chromatin remodeling at estrogen receptor (ER), aromatase, and AR promoters (males > females), particularly in the sex dimorphic regions, is dimorphic as well (Matsuda et al. 2008; Matsuda et al. 2012; Murray et al. 2009; Tsai et al. 2009); (3) sex differences in histone 3 methylation and acetylation, in parallel with testosterone surge, cause a dimorphism in the overall transcriptional activity in selected brain areas (males > females; Tsai et al. 2009). In addition, the activated sex steroid hormone receptors associate with histone modifying enzymes, modulating chromatin remodeling (Gao and Alumkal 2010; Leader et al. 2006) and, concomitantly, AR transcriptional activity is potentiated by the association with histone demethylases (Casati et al. 2013). This bidirectional interaction between sex steroid hormones and the epigenetic mechanisms early in life might be an important cue to determine and to maintain a dimorphic phenotype throughout the lifespan.
It is evident that interfering with the complex modulation of this highly regulated dimorphic process during the sensitive developmental window may cause long-lasting effects, and that BPA, behaving both as a estrogen (Delfosse et al. 2014) and as an antiandrogen (Picot et al. 2014; Teng et al. 2013), may have a powerful influence on most of these processes.
A huge body of data demonstrate that developmental exposure to low BPA interferes with the brain sex differentiation process with enduring effects on brain structure and functions. However, few articles analyzed specifically whether the molecular targets were other than the variation of ER and/or AR activation. Among the actors possibly involved (sex steroid receptors and testosterone activating enzymes), the majority of the available data examined how BPA influences the expression pattern of ERα and ERβ both in hypothalamic and extra-hypothalamic brain areas (Cao et al. 2013; Picot et al. 2014). Overall, the variation of ER expression levels in the 2 sexes affects the physiological dimorphism in nuclear morphology and functions (Cao et al. 2014; Rebuli et al. 2014, and the section below). In some cases, this results in a male showing a more feminine phenotype and vice versa, while in other cases the loss of dimorphism is due to a change only in 1 of the 2 sexes (see Richter et al. 2007 and below for further references). Sometimes, the observed outcomes are not completely reproduced by the exposure to estrogens (Rebuli et al. 2014), suggesting that BPA does not act only as estrogen pure agonist on some parameters. Still, it should be mentioned that other authors did not find alteration of the morphology and of ERα expression levels in the dimorphic hypothalamic nuclei after developmental exposure (Picot et al. 2014).
Another important player in brain sex development, at least in rodents, is the aromatase enzyme (see above); the very few data present in the literature demonstrate that BPA perinatal exposure stimulates dose dependently brain hippocampal aromatase expression in male animals only in the early postnatal period (Xu et al. 2010); the restriction of BPA effect on aromatase to the first week of postnatal life is not surprising, since in normal conditions, the expression of this enzyme drastically decreases afterward (Colciago et al. 2009). It is interesting to note that BPA also induces a strong overexpression of the brain-specific aromatase in some subregions of the developing zebrafish brain through an ER-mediated mechanism (Chung et al. 2011). Though, early BPA exposure might interfere with the process of brain sex differentiation also controlling the endogenous availability of estrogens.
Regarding the androgen-AR system, no data are present in the literature on BPA influence on the expression of the two 5α-reductases, and it seems that AR expression is increased in male hypothalamic SDN-POA only after adult exposure (Picot et al. 2014). Yet, more studies are needed to clarify the possible influence of low BPA dose on these important developmental parameters since a significant decrease in the expression levels of both 5α-reductases and AR have been found after chronic exposure of adult rat in the prostate (Sanchez et al. 2013) and in the seminiferous epithelium (Qiu et al. 2013).
The recent evidence that BPA also modifies the DNA methylation degree of ER gene promoters (disrupting the epigenetic programming of their expression) in a dose-, sex-, and region-specific manner (Kundakovic et al. 2013) further underlines the enduring disruptive action of BPA on the determination of the brain dimorphic phenotype and supports the concern about the exposure to this endocrine disruptor (EDC).
Neuroendocrine Targets
The neuroendocrine hypothalamus is the main interface between environment and body, controlling basic homeostatic responses. Hypothalamic neurons respond to inputs coming from periphery, from the rest of the CNS, and from the environment through the secretion of releasing hormones (RHs), of neurohormones/neuropeptides, and through the activation of autonomic responses. Typically, RHs act at the anterior pituitary gland controlling the release of corresponding tropins, which stimulate the hormonal production from peripheral endocrine glands; peripheral hormones, activating their cognate receptors, control RH and tropin secretion by negative feedback mechanisms. Perinatal as well as adult exposure to BPA alter several aspects of the neuroendocrine axis functions acting both at central (hypothalamus and/or pituitary) and peripheral (endocrine gland) levels. This section refers specifically to the effects of BPA on hypothalamic neuroendocrine functions.
Thyroid axis
Thyroid hormones are essential for normal brain development and prenatal disruption or inhibition of the thyroid axis causes deep deficiencies in adult brain functions. Even though BPA binds to thyroid hormone receptors (TRs) with low affinity, suggesting that high levels are required to disrupt this axis, some in vitro studies revealed that low BPA inhibits TR-mediated gene expression enhancing the recruitment of corepressors (discussed in Rubin 2011). Notably, developmental exposure to low BPA in rats produces an endocrine profile similar to that observed in thyroid resistance syndrome. This syndrome is characterized by the inhibition of TR-mediated negative feedback at the hypothalamus/pituitary level (resulting in the increase of circulating thyroid hormones) and by the increase of thyroid-responsive gene expression in the brain (Itoh et al. 2012; Zoeller et al. 2005). The TR-antagonistic activity of BPA seems to affect also specific developmental events controlled by thyroid hormones such as oligodendrocyte differentiation from precursor cells (Seiwa et al. 2004). As opposed, the commonly used alogenated derivatives of BPA are more active on the thyroid axis and may behave as TR agonists or antagonists, depending on the type of TR present (see Rubin 2011 for references).
Adrenal axis and stress response
The hypothalamo–pituitary–adrenal (HPA) axis has an essential role in the neuroendocrine control of stress responses, and modification of its functions during development has been linked to many neuroendocrine disorders and cognitive deficits (Panagiotidou et al. 2014). In silico and in vitro studies indicate that BPA can bind to and activate glucocorticoid receptors (GRs) with an affinity similar to natural or synthetic glucocorticoids (see Rubin 2011 for references). Moreover, it is well known that both the development of brain nuclei directly involved in stress responses (eg the locus ceruleum, Kubo et al. 2001) and the HPA axis are sensitive to sex steroid hormones (Viau 2002). Thus, BPA might interfere with this neuroendocrine system both directly and by modulating sex hormone activity. Rats prenatally exposed to low BPA show morphofunctional alteration of the adrenal glands, which results in the increase of basal and stress-stimulated glucocorticoid secretion in adulthood (Poimenova et al. 2010). In addition, exposed animals show reduced brain GR levels in comparison to controls and an altered response to stress conditions (Panagiotidou et al. 2014). However, it should be mentioned that the opposite has been described by other authors (Chen et al. 2014), who reported an increase of hypothalamic GR expression, coupled to an elevated activity of all the components of the axis. Of interest is that the 2 articles agree on the dimorphic response of exposed animals to stress, being males more susceptible than females. Notably, prenatal exposure to BPA at higher doses (1.5 mg/kg/d) alters the sexual differentiation of the locus coeruleus (Kubo et al. 2001). These few conflicting results indicate that a clear picture of the specific BPA effects on HPA function is still lacking; due to the involvement of the axis in stress responses, this should be an important area for future research.
Reproductive axis and sex behavior
At present, there is a high concern on EDC exposure and human reproductive health (Peretz et al. 2014). Growing evidence from research on laboratory animals and humans supports the view that BPA adversely affects both male and female reproductive functions not only interfering with gonadal development and gonadal hormone functions (see other articles of this special issue), but also with the development and activity of the hypothalamic gonadotropin releasing hormone (GnRH) neurons. These neurons, located in the arcuate nucleus and in the POA, release the GnRH, which controls gonadotropin secretion from pituitary gland. The GnRH neurons represent a unique class among the hypothalamic neuroendocrine cells, because during embryogenesis they develop in the olfactory placode and migrate along vomeronasal nerves to the hypothalamus, where it undergoes terminal differentiation (Cariboni et al. 2007). All these steps are under the strict control of several developmental cues, and the interference with their action might represent a cause of important reproductive disorders, such as Kallmann syndrome (Cariboni and Maggi 2006). The ability of BPA to interfere with embryonic GnRH neuron migration is under investigation in our laboratory using the immortalized immature murine GnRH neurons (GN11 cells), which possess migratory activity (see Cariboni et al. 2007 for more information) and express of both ERα and ERβ (Ng et al. 2009). Preliminary data clearly indicate that in vitro exposure to nanomolar concentration of BPA stimulates GN11 migration dose dependently; this effect is significantly reduced by the pure antagonist ICI 182 780, suggesting the direct involvement of ER activation (Galbiati M and Negri-Cesi P, unpublished). Our results agree with the ability of BPA to stimulate cancer cell migration (Derouiche et al. 2013; Ptak et al. 2014) and is in line with the influence BPA exerts on neuronal migration during embryogenesis (see Basic Mechanisms of Brain Development section).
The frequency of GnRH pulsatile secretion, which is critical for luteinizing hormone (LH) release from the pituitary gland, is modulated by the activity of AVPV neurons (Yeo 2013). Main components of the AVPV network are the stimulatory peptide Kisspeptin1 (Kiss) and the 2 inhibitory peptides (transcriptional regulator enhanced at puberty [EAP1] and peptide YY [YY1]). It is important to recall that AVPV is a dimorphic nucleus, larger in females than in males (see above); furthermore, kisspeptin neurons innervating GnRH cells are less abundant in males than in females, due to the prenatal specific effect of testosterone-derived estrogens (Clarkson and Herbison 2006). Both in rodents and humans, the integrity and the dimorphic activity of this complex system are essential for puberty onset in both sexes and for the gender-specific LH secretion (tonic in males, cyclic in females; reviewed by Mueller and Heger 2014). Indeed, variation of GnRH pulsatility triggers puberty onset and is fundamental for the mid-cycle LH-surge, which drives ovulation. Recent in vitro studies demonstrate that short lasting treatment with BPA stimulates Kiss, while inhibiting EAP1 and YY1 expression with a mechanism mediated by ERα activation (Mueller and Heger 2014). Thus, BPA by perturbing the balance between stimulatory and inhibitory inputs to GnRH neurons may cause the up-regulation of GnRH release. The BPA-induced GnRH hypersecretion from female rat neurons has been demonstrated also in an in vitro perfusion system (Gore 2010). In line with the in vitro studies, in vivo neonatal exposure to low BPA seems to permanently affect GnRH pulsatile release in female rats, resulting in the acceleration of puberty onset and in altered estrous cyclicity (Fernandez et al. 2009).
The BPA prenatal exposure also modifies AVPV morphology causing the masculinization of the nucleus in females (decreased size) and the demasculinization in males (Bai et al. 2011; McCaffrey et al. 2013; Patisaul et al. 2006). In particular, BPA-treated males possess more Kiss-expressing cells in the AVPV (Bai et al. 2011) and display a stable, female-like, LH surge when estrogen primed (Bai et al. 2011).
The BPA seems to affect also SDN-POA morphology and function in males, exerting a demasculinizing action on the nucleus. This is confirmed by the reduced number of calbindin-expressing neurons, a reliable marker of SDN-POA borders (McCaffrey et al. 2013). Conversely, BPA is unable to either masculinize the nucleus in females (McCaffrey et al. 2013) or to induce mounting behavior in adult ovariectomized animals primed with testosterone (Naule et al. 2014); rather, it exacerbates the female copulatory behavior, increasing the lordosis quotient in the same animals (Naule et al. 2014). As reviewed by Wolstenholme and coworkers (2011), contrasting results are available in the literature on the effect of prenatal BPA exposure on female sexual behavior. While some authors did not found changes in lordosis quotient in exposed animals (Ryan et al. 2010), others reported an increased sexual motivation and receptivity (Farabollini et al. 2002). Notably, the exposure of juvenile females to the same regimen causes a defeminization of social interactions and social grooming (Porrini et al. 2005), coupled to the masculinization of play behavior (Dessi-Fulgheri et al. 2002). The impact of BPA on the sexual behavior of exposed male animals—intromission frequency and latency to intromission—is positive or negative depending on the time of exposure—prenatal or postnatal—(Farabollini et al. 2002).
In conclusion, it is clear that BPA disrupts the dimorphic function of the AVPV and SDN-POA networks; conversely, uncertainty still remains on the final outcome in female and male reproductive physiology and sexual behavior. As outlined in the Introduction section, this inconsistency is possibly due to differences in the duration and extent of the exposure, as well as of the route of administration.
It is important to emphasize that as androgens rather than estrogens are central for prenatal brain masculinization in humans (see the previous section), it would not be correct to apply findings obtained in rodents in human risk assessment. Though, the epidemiological association of early human exposure with precocious puberty in girls and with declining sexual desire in men, described by some authors (Manfo et al. 2014; Rochester 2013), strongly suggests an adverse effect on the neuroendocrine control of the reproductive axis and sex behavior also in humans and stresses the urgent need of more and more controlled basic research to clearly define risk assessment in human beings.
Effect on the hypothalamic control of food intake and energy expenditure
In physiological conditions, complex homeostatic processes control the maintenance of energy balance to a predetermined set point. These processes are able to detect nutrient storage and energy levels, to integrate this information and to coordinate food intake and energy expenditure accordingly. A distinct circuitry of interconnected neurons located within the hypothalamus (in the arcuate nucleus, in the dorsomedial hypothalamus, and in the lateral hypothalamic area) perceive and integrate all the peripheral and central information related to the nutritional status (circulating nutrient and hormones levels, autonomic inputs) and respond modifying neuronal firing and/or secreting orexigenic (mainly neuropeptide Y [NPY] and Agouti related peptide [AgRP]) or anorexigenic (mainly proopiomelanocortin [POMC] and cocaine-amphetamine regulated transcript [CART]) neuropeptides (Parker and Bloom 2012; Saper et al. 2002). These neuropeptides interact with specific membrane receptors and control both their own secretion reciprocally and the activity of other brain centers (Parker and Bloom 2012), adjusting food intake in response to changing energy requirements. Among peripheral hormonal signals, leptin (from adipose tissue) and some gut hormones (eg ghrelin and cholecystochinin) modulate estrogenic and antiandrogenic neuropeptide secretion in opposite ways, controlling food intake and energy expenditure over the short (ie meal size) and the long (ie energy stores) periods. For a recent overview of the central mechanisms regulating feeding behavior see Sobrino Crespo et al. 2014.
The activity of this neuronal circuitry is gender specific, with females showing responsiveness to various anorectic inputs different from that of males (Mackay et al. 2013). This dimorphism arises from the organizational action of sex steroids during development (see above), as suggested by the impairment of the anorectic response to leptin and by the development of male-like energy balance circuitry in females neonatally exposed to testosterone (Nohara et al. 2011). Since the masculinization of the female POMC circuitry can be mimicked by neonatal DHT administration (Nohara et al. 2011), it seems that androgens rather than testosterone-derived estrogens are involved in the dimorphic differentiation of the hypothalamic feeding circuits. Notably, the neonatal exposure to testosterone-derived estrogens predisposes females to hyperleptinemia, leptin resistance, and obesity (Nohara et al. 2011). Thus, the disruption of gonadal hormone action during development may lead to the alteration of the dimorphic feeding behavior.
Sex steroid hormones, particularly estrogens, can influence the activity of this plastic network also in adult animals; indeed, estrogen administration has ER-mediated anorectic properties (Liang et al. 2002) and increases excitatory inputs to POMC neurons in the arcuate nucleus with a resulting decrease in food intake (Gao et al. 2007). Estrogens can also modulate the production of NPY and AgRP (Titolo et al. 2006); the ability of the hormone to activate or to repress the orexigenic neuropeptide transcription depends on the ERα/ERβ status: while the activation of both ERα and ERβ is required for the repression of the 2 neuropeptide production, ERβ alone is essential for their induction (Titolo et al. 2006).
In addition to genetic predisposition and to the in utero nutritional status, also the perinatal exposure to EDC is a critical factor contributing to the etiology of nutritional pathologies (Grun and Blumberg 2009; Schneider et al. 2014). Acting both at genetic and at epigenetic levels, these influences seem to modify the physiological programming of energy homeostasis with an imprinting mechanism that becomes evident much later in life (Dyer and Rosenfeld 2011). Even though not all the authors agree (Sharpe and Drake 2010), there is now increasing concern on the involvement of early-life exposure to low BPA in the onset of human obesity and related metabolic complications (Fenichel et al. 2013; Le Corre et al. 2013).
The present research on the relationship between BPA and dismetabolism focuses almost exclusively on adipose tissue development and functions after perinatal or adult exposure. It has been shown both in vitro and in vivo (in experimental animal models or humans) that the obesogenic activity of BPA relies on its ability to accelerate adipogenesis (increasing preadipocyte differentiation and the expression of adipogenic genes), to induce lipid accumulation, to modify the metabolic functions of the adipose tissue, and to influence adipokine release (see Ben-Jonathan et al. 2009; Le Corre et al. 2013; Rubin 2011; Vom Saal et al. 2012). The alteration of adipokine secretion from adipose tissue, caused by a constant BPA exposure both in animals and in humans, might contribute to the disruption of the response of the hypothalamic circuitry controlling energy balance to these peripheral signals.
Whether BPA directly affects the hypothalamic network regulating food intake and energy expenditure is still unclear, but some authors demonstrate that BPA at micromolar dose either intracerebroventricular (ICV) administered to newborn rats (Masuo et al. 2004) or after in utero/early postnatal exposure (Mackay et al. 2013) affects the expression of some selected orectic and anorectic neuropeptides in adulthood. Moreover, BPA seems to exert an organizational effect on the neuronal network, both masculinizing the gene expression profile and influencing how the circuitry respond to dietary challenge. Thus, low BPA exposure during early life might lead to a gender-specific adverse metabolic phenotype and should be considered as a risk factor involved in the developmental origin of metabolic diseases. As previously mentioned, the estrogen-ER (α/β) system modulates the activity of NPY and AgRP neurons also in adult animals (Titolo et al. 2006). Hence, the possible interference of adult BPA exposure with estrogen action on feeding circuitry might potentiate the obesogenic activity of the compound.
The study on the epigenetic alterations in the expression of genes involved in the central control of food intake by environmental factors is relevant, but very few data have been provided on this topic so far. Among these, prenatal undernutrition reduces POMC levels in the rat brain by gene hypermethylation (Stevens et al. 2011); POMC hypermethylation might be linked to childhood obesity (Kuehnen et al. 2012); hypomethylation of the hypothalamic satiety receptor melanocortin-4 receptor (Mc4 R) gene seems to be related to high fat diet in mice (Widiker et al. 2010). It is known that developmental exposure to BPA causes epigenetic reprogramming (Ryan et al. 2010; Bernal and Jirtle 2010) and has a genome wide effect on the DNA methylation degree within the brain (Kundakovic and Champagne 2011; Kundakovic et al. 2013); however, to the author’s knowledge, the epigenetic influence of BPA on the battery of genes involved in the hypothalamic control of energy balance has never been evaluated.
Effect on Other Brain Region and Functions
Many nonreproductive dimorphic behaviors are affected by BPA. It has been indeed demonstrated that prenatal exposure to low doses abolishes or reverts the dimorphism in novelty response behavior and locomotor activity of adult animals, paralleled by an estrogen-dependent modification of the size of the brain region controlling these behaviors (Kubo et al. 2003).
A number of reports have shown that BPA developmental exposure strongly affects anxiety-related behaviors in rodents, often blunting or reversing the normal dimorphic response in classical anxiety tests such as the elevated plus maze (Patisaul and Bateman 2008; Rubin et al. 2006) and the exploration in open-field (Gioiosa et al. 2007; Kubo et al. 2003). Notably, the increased anxiety shown by exposed females in exploratory tests goes in parallel to higher basal and stress-stimulated corticosterone levels (Poimenova et al. 2010). Taken as a whole, it appears from these and other behavioral evidences (see Wolstenholme et al. 2011 for references) that BPA might affect anxiety either interfering with the estrogen-induced sexual differentiation of the brain or via alteration of the HPA axis.
Memory formation and utilization is a dimorphic and complex process involving the hippocampus and several other brain structures. A thorough description of the molecular mechanisms of memorization is beyond the purposes of this article; however, basically, they involve glutamatergic system activation and hippocampal synaptic remodeling. In line with the potent influence of estrogens and other hormonal steroids on hippocampal development and on its functions in adulthood, developmental exposure to BPA has a long-lasting effect, impairing learning in many spatial and nonspatial memory tests (see Kundakovic and Champagne 2011 and Wolstenholme et al. 2011 for references); females seem to be more sensitive than males to BPA interference in some tests. Some of the described changes are associated with decreased expression of some subunits of the N-methyl-D-aspartate (NMDA) glutamatergic receptors in the hippocampus (Xu et al. 2010), as well as to modification of the synaptic density and structure (Wang et al. 2014; Xu et al. 2013), suggesting a neurobiological substrate of BPA-induced cognitive dysfunction. It should be mentioned that some authors found that BPA exposure does not affect the cognitive performance (Sadowski et al. 2014).
Concluding Remarks
The potential detrimental actions of BPA on brain differentiation and functions are multiple and extremely complex, as they may involve different hormonal and epigenetic pathways, each one activated, possibly in a dimorphic way, during a specific window of susceptibility. The resulting changes may then lead to altered reproductive and nonreproductive behaviors.
In spite of the huge amount of literature in these last years, controversies still remain on the actual safety of a prenatal exposure to low dose BPA. This uncertainty stems from discrepancies in the outcomes obtained in the different laboratories caused, among others, by different experimental designs, route and window of exposure, and experimental targets. Nonetheless, the current evidence is enough to raise concern for human health and warrants to adopt a precautionary principle, particularly to protect a developing organism. The assessment of BPA brain accumulation in relation to the extent of the exposure and to the appearance of the adverse effects in future basic experimental studies will certainly give more precise indication to be translated into dose–response models and in a better definition of the warning threshold of human exposure.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.