
Abstract
Polyacrylonitrile-based pre-oxidized fibers with improved thermal stability, flame retardant, and mechanical properties were made from the pristine polyacrylonitrile fibers through chemical pretreatment followed by pre-oxidation in air. The morphological structure of the polyacrylonitrile-based pre-oxidized fibers was investigated by Fourier transfer infrared spectra, X-ray diffraction, scanning electron microscopy, and X-ray energy dispersive spectrometer. The changes of characteristic functional groups and chemical compositions confirmed the successful modification of the polyacrylonitrile fibers during pre-treatment. The grooves and cracks on the surface of polyacrylonitrile-based pre-oxidized fibers were remarkably decreased in comparison with that of pristine polyacrylonitrile fibers. The evolution of crystalline structure of the polyacrylonitrile fibers proved the occurrence of cyclization reactions during pre-oxidation. Meanwhile, thermal stability, flame retardant, and mechanical properties of polyacrylonitrile-based pre-oxidized fibers were also investigated by thermogravimetric analyzer, oxygen index meter, micro combustion calorimeter, and single fiber tensile tester, respectively. The results demonstrated that the polyacrylonitrile-based pre-oxidized fibers initially pre-treated by hydroxylamine hydrochloride, followed by monoethanolamine, had a high limiting oxygen index of 40.1 and breaking strength of 2.03 cN/dtex. The peak of heat release rate and total heat release of polyacrylonitrile-based pre-oxidized fibers decreased significantly while its charred residues increased, contributing to the improved flame retardant property.
Introduction
The pre-oxidized polyacrylonitrile fibers (OPF) are novel flame retardant fibers, which are obtained by thermal-stable treatment of the polyacrylonitrile (PAN) fibers in air atmosphere.1–3 During pre-oxidation stage (a lower-temperature thermal stabilization stage of about 190ºC–300ºC), it is generally agreed that three chemical reactions occur in the fibers: cyclization, dehydrogenation, and oxidation.4,5 This produces a large amount of heat and low-molecular by-products, which cause a significant change in the fibers structure and then leads to the formation of a ladder-type structure.6–10 Compared with other flame retardant fibers, in addition to safety, good processability, and comfort, the OPF also has the characteristics of high-temperature resistance, good flame retardant property, and low cost.11,12 In recent years, it has been widely used in construction, military, aerospace, automobile, and other fields.
A large amount of heat generated by the PAN fibers during pre-oxidation leads to poor and low elongation, which has become one of important factor of affecting the structure and property of the OPF.13–15 PAN contains at least 85% of acrylonitrile monomer on macromolecular main chains, and the cyano (C≡N) groups can participate in various chemical reactions.16,17 Therefore, chemical pre-treatment of the PAN fibers followed by pre-oxidation in air can improve chemical structure and obtain pre-oxidized fibers with excellent properties.18–20 Aiming at the chemical pre-treatment of PAN fibers, initially hydroxylamine (HA) hydrochloride is widely used as a modification reagent, followed by hydrazine hydrate as a crosslinking agent.21–23 They are effective modification methods to increase heat resistance and thermal stability of PAN fibers. However, with the concept of green chemistry rooted in the heart of people, reactions performed in efficient, inexpensive, halogen-free, and low-toxic solvents with obvious advantages of low-cost and minimal environmental impact have received much attention. There are numerous literatures reporting the pre-treatment of the PAN fibers before pre-oxidation but most of the chemicals used are directly utilized to prepare carbon fibers.24–28
To the best of our knowledge, the effects of chemical pre-treatment on structure and property of PAN-based pre-oxidized fibers have not been reported. Therefore, in this study, a simple and convenient impregnation method was proposed to treat the pristine PAN fibers; in other words the OPF were made from the pristine PAN fibers through pre-treated with HA hydrochloride and monoethanolamine (MEA) alone or jointly followed by pre-oxidation in air. The structural morphology, thermal stability, flame retardant, and mechanical properties of OPF were emphatically investigated.
Experimental
Materials
PAN fibers, a commercial product, were kindly provided by Jilin chemical industrial company Ltd., China. HA hydrochloride, MEA, anhydrous sodium carbonate, and alcohol (all from Sinopharm chemical reagent Co. Ltd., Shanghai, China) were analytically pure and used as received.
Chemical pre-treatments of PAN fibers
PAN fibers were first washed with alcohol to remove oil traces and impurities on the fiber surface. A certain concentration of HA (40 g/L) and MEA (30 mg/L) solution was configured. First, the pristine PAN fibers was pre-treated in the HA aqueous solution at 80°C for 2 h, and adjusted the solution pH value to 5 with anhydrous sodium carbonate, then washed, dried, and prepared the PANHA fibers. Then, the PANHA fibers were added into a certain concentration of MEA solution. The reaction mixtures were refluxed at 90ºC under stirring for 2 h. The reaction product was cooled to room temperature and then washed with deionized water at 70ºC to gain PANHA-MEA fibers. To investigate the effects of different chemicals and its pre-treatment sequence on structural morphology and property of OPF, we also prepared other various modified fibers of PANHA, PANMEA, and PANMEA-HA with similar processes. The schematic illustration of chemical reactions during pre-treatment was presented in Figure 1.

Schematic illustration of chemical reactions during pre-treatment.
Preparation of the OPF
A certain tension was applied to the modified PAN samples, which were placed in a muffle furnace and subjected to elevated temperature pre-oxidation. The pre-oxidation starting temperature was 170ºC with the heating rate of 1°C/min, the temperature was raised to 250ºC and maintained for 10 min, to obtain the OPF.
Measurements
The Fourier transfer infrared (FT-IR) spectrometer was carried out with a Nicolet 10 (American Thermo Fisher Scientific Co. Ltd., China) spectrometer. The wavenumber resolution was 2 cm−1 and the scan region was from 4000 to 400 cm−1. The changes in surface morphology were observed with a scanning electron microscope (SEM, SU 1510, Hitachi Co. Ltd., Japan). X-ray energy dispersive spectrometer (EDS) was simultaneously conducted with SEM measurements. X-ray diffraction (XRD) measurements were taken by an X-ray diffractometer (D2 Phaser, Bruker AXS GmbH, Germany). And the 2θ angle ranged from 5º to 40º. The mechanical property was tested using a Single fiber tensile tester (YG(B) 026D-250, Wenzhou Darong textile standard instrument Co. Ltd., China). The samples were conducted at a stretching speed of 20 mm/min with a gauge length of 20 mm. Finely powdered fiber samples were used for thermogravimetric (TG) analysis with a TA instrument Q-500. The measured temperature was varied from 30ºC to 700ºC with the heating rate of 10ºC/min under nitrogen atmosphere. Limiting oxygen index (LOI) measurement was carried on an LOI analyzer (JF-3, Beijing Xinsheng excellence technology Co. Ltd., China), and the samples were made by twisting fibers according to the FZ/T 50017-2011 standard. Flame retardant property was characterized by micro combustion calorimeter (MCC) based on the principle of oxygen consumption. The signals from the MCC were recorded and analyzed by a computer. The dried fiber samples were first pyrolyzed with a linear heating rate of 1ºC/s in the furnace fixed with MCC. The decomposed components were carried away by an inert gas; subsequently, they were mixed with oxygen followed by being combusted at 900ºC. The mass of samples for the MCC tests was about 4–6 mg.
Results and discussion
Chemical structures
The FT-IR spectra of PAN fibers treated with HA and MEA alone or jointly were presented in Figure 2. It could be seen that the PAN fibers had the stretching vibrations of the C–H group absorption peak at 2939 and 2850 cm–1. The bands centered at 2243 cm−1 and 1732 cm−1 were attributed to the stretching vibrations of cyano (C≡N) and ester (C=O) groups, respectively. After chemical pre-treatment, the intensity of characteristic peak of the C≡N groups at 2243 cm−1 was significantly decreased and the occurrences of new absorption bands located at about 1647 and 911 cm−1 were assigned to the stretching vibrations of C=N and N–O groups. The band centered at 1593 cm−1 was due to the stretching vibrations of C–NH2 in the amidoxime groups. 28 The results indicated that there were chemical reactions between C≡N groups of PAN fibers and chemical regents (HA and/or MEA). For the FT-IR spectra of PANMEA-HA and PANHA-MEA, the strength of the C≡N characteristic peak was further decreased to almost completely disappearance, while a new peak at ~1560 cm−1 occurred due to the overlaps of the stretching vibrations of C–N groups and the bend vibrations of N–H groups. The changes of the characteristic peaks indicated that the mixed solutions of HA and MEA could convert more C≡N groups of PAN fibers to the amide groups than individual HA or MEA. Meanwhile, in the spectrum of PANHA-MEA, the characteristic peak of C≡N groups at 2243 cm−1 significantly weakened or disappeared, indicating the conversion degree of C≡N groups for PANHA-MEA was higher than that of PANMEA-HA. The reasons might be that the length of the grafted molecular chain would affect the polarity of the functional groups to a certain degree, and the steric hindrance generated by the existing molecular chain would affect the subsequent grafting reactions. Besides, it could be observed that the adsorption peaks of C=O in amide groups overlapped with the bending vibrations of N–H groups and the stretching vibrations of C=C groups in the range of 1500–1700 cm−1. All of the above results showed the amide and amino groups were incorporated into the PAN fibers.
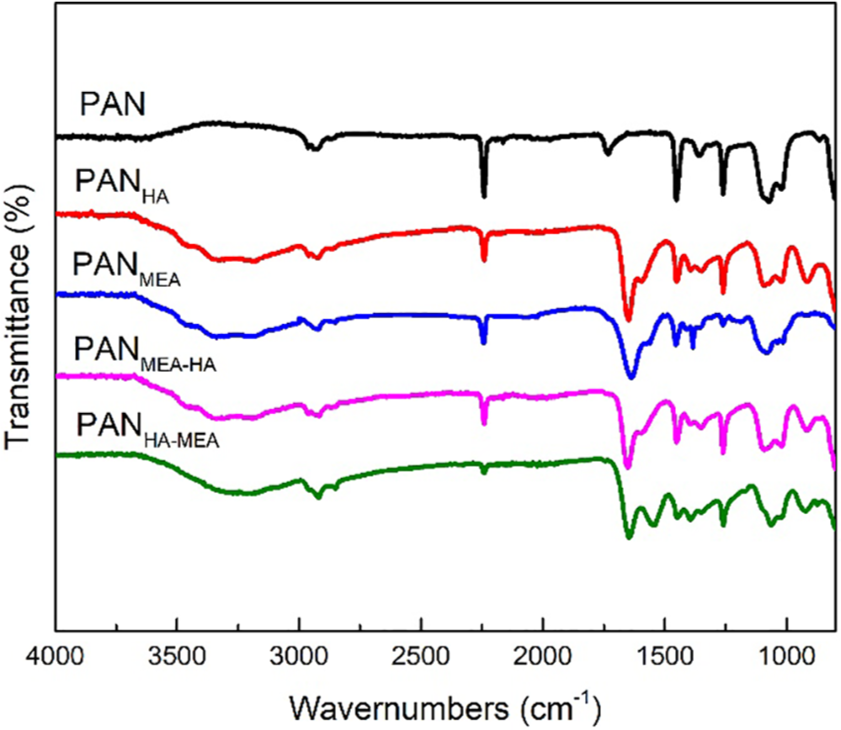
FT-IR spectra of PAN fibers before and after chemical pre-treatment.
In order to further prove the success of the modification, the changes of chemical compositions of PAN fibers was observed by EDS, and the acquired results were summarized in Table 1. As we could see, the content of the C element for PAN fibers pre-treated by HA or MEA was decreased, while the N content of PAN fibers was increased. Meanwhile, the PAN fibers pre-treated by HA (i.e. PANHA) had a larger N/C ratio because it contained more N elements per unit mass fraction. Compared with PANHA and PANMEA pre-treated by an individual HA or MEA solution, the content of the C element for PANMEA-HA and PANHA-MEA was relatively increased after the pre-treatment combined with HA and MEA. This was caused by the introduction of the long carbon chains in the MEA. Moreover, the final N/C ratios for all PAN fibers after chemical pre-treatments were increased, which gave strong evidence that the functional groups had been grafted with the macromolecular structures of PAN fibers. In addition, the different N/C ratios between PANMEA-HA and PANHA-MEA could be also observed. This might be because the carbon chains of the MEA was longer than that of the HA. When PAN fibers was first pre-treated by MEA, the carbon chains grafted had a certain spatial hindrance effect, which limited the subsequent modification of the fibers by the HA. This result was consistence with the analysis of FT-IR spectra (i.e. PANHA-MEA with high conversion degree of C≡N groups).
Chemical compositions of PAN fibers obtained from EDS in %.

EDS: energy dispersive spectrometer; PAN: polyarylonitrile; PANHA: hydroxylamine treated polyarylonitrile; PANMEA: monoethanolamine treated polyarylonitrile; PANMEA-HA: monoethanolamine and hydroxylamine treated polyarylonitrile; PANHA-MEA: hydroxylamine and monoethanolamine treated polyarylonitrile.
Structural morphology
Figure 3 demonstrated the representative SEM micrographs of pristine PAN fibers and OPF with and without chemical pre-treatments (i.e. OPF, OPFHA, OPFMEA, OPFMEA-HA, and OPFHA-MEA). Figure 3(a) showed that the surface of pristine PAN fibers was covered with cracks and grooves due to the formed micro-defect structures during manufacturing process. The surface of the pre-oxidized fibers (without chemical pre-treatment) formed some shallower cracks, as shown in Figure 3(b). This was because the fibers’ surface not only increased the density of the macromolecular structure during the pre-oxidation process, but also caused a certain degree of oxidative ablation. These led to a slight change on fibers’ surface. After chemical pre-treatment and pre-oxidization, the HA and MEA pre-treated OPF had a relatively smooth surface compared with the pre-oxidized fibers (i.e. OPF), as shown in Figure 3(c) to (f). The HA might have a catalytic effect, which prevented the fibers from generating a concentrated heat release in the later stage of pre-oxidation and so prevented fibers’ defects.

SEM images of (a) pristine PAN fibers, (b) OPF, (c) OPFHA, (d) OPFMEA, (e) OPFMEA-HA, and (f) OPFHA-MEA.
The X-ray diffraction was used to characterize the crystalline structure of OPF, the XRD patterns were illustrated in Figure 4. The diffraction peaks centered at 2θ angles of 17º and 29º corresponded to (100) and (110) crystallographic planes of the pristine PAN fibers, respectively. 29 After pre-oxidation, the peak position centered at 17º was not shifted while the intensity significantly weakened, indicating that the PAN macromolecular chains were tended to be disordered. A new diffraction peak appeared at 25º indicated that the original sequence structure was gradually destroyed and a new sequence structure was gradually formed. This was because PAN macromolecular chains were cyclized via dehydrogenation and formed a six-membered heterocyclic structure with a large d-spacing. Therefore, the area of the peak at 2θ = 25º could reflect the gradual accumulation of the cyclized structure formed by the PAN molecules during pre-oxidation. After chemical pre-treatment and pre-oxidization, the diffraction peak at 17o decreased and even disappeared while the diffraction peak at 25º widened. Combined with FT-IR analyses, we knew that the conversion rate of C≡N groups of PAN fibers was different through different chemical pre-treatments. This would affect the content of the stacked and/or cyclizing structures. At the same pre-oxidation conditions, the degree of cyclization of the OPF through chemical pre-treatment with HA followed by MEA (i.e. OPFHA-MEA) was higher, which would inevitably affect the flame retardant and mechanical properties of OPF.

XRD patterns of the OPF with and/or without chemical pre-treatment.
Mechanical property
The effects of different chemical pre-treatments with HA and/or MEA on the mechanical property of OPF were carried out, and the experimental results were listed in Table 2. During the pre-oxidation process, both the reduction of polar groups and the breakage of a part of the macromolecular backbones led to a decrease in the coherent energy of the fibers; thus the fibers’ strength happened to decrease. After chemical pre-treatment with HA or MEA alone, the pre-oxidation reaction temperature of the fibers decreased. In other words, the pre-oxidation reactions occurred at a lower temperature for the PAN fibers, so the temperature of the oxidative exothermic reactions became wide and the concentrated exothermic phenomenon was alleviated. The damage of the concentrated exotherms on the mechanical property of the fibers was relieved, hence the obtained pre-oxidized fibers had better mechanical property. As we could see from the Table 2, the obtained OPF pre-treated by HA and MEA (i.e. OPFHA-MEA and OPFMEA-HA) demonstrated superior mechanical property in comparison with those OPF pre-treated by HA or MEA alone (i.e. OPFHA and OPFMEA). The performance of OPFHA-MEA was also better than that of OPFMEA-HA. The breaking strength and breaking elongation of the OPFHA-MEA fibers reached 2.03 cN/dtex and 19.1%, respectively. It was speculated that the chemical pre-treatment combined with HA and MEA might introduce more amide groups into the PAN macromolecular side chains, which promoted the cyclization reactions and formed a stable cyclic pyridine structures. At the same time, it avoided the fibers’ damage caused by the concentrated exothermic heat in the late stage of pre-oxidation. The above two aspects contributed to high mechanical property of the obtained OPF.
Limiting oxygen index values and mechanical property of OPF with and/or without chemical pre-treatment.

LOI: limiting oxygen index; OPF: pre-oxidized polyacrylonitrile fibers; OPFHA: hydroxylamine treated pre-oxidized polyacrylonitrile fibers; OPFMEA: monoethanolamine treated pre-oxidized polyacrylonitrile fibers; OPFME-AHA: monoethanolamine and hydroxylamine treated pre-oxidized polyacrylonitrile fibers; OPFHA-MEA: hydroxylamine and monoethanolamine treated pre-oxidized polyacrylonitrile fibers.
Thermal stability
The thermal stability of OPF was studied using thermogravimetric analysis in an N2 atmosphere, and the corresponding TG curves were shown in Figure 5. It could be found from Figure 5 that all OPF samples demonstrated the same degradation trends. The temperature at 5% weight loss of the OPFHA-MEA was 104ºC, which was not much different from that of other OPF. The corresponding weight loss was caused by moisture adsorbed and small molecule functional groups. When the temperature reached up to 400ºC, the weight loss of OPF suddenly increased. This was due to the presence of more oxygen atoms in the pre-oxidized fibers, which decomposed to form small molecular oxides at higher temperatures. At 800ºC, compared with the charred residues of 56 wt% for the OPF, the charred residues of the OPF via chemical pre-treatment were significantly increased. The charred residues of the OPFHA-MEA reached up to 68 wt%. The reasons might be that after the chemical pre-treatments, the exothermic reactions of the fibers were more moderate and the thermal degradations of the fibers were relatively reduced. Moreover, the combined pre-treatment with HA and MEA promoted the stability of the C–N conjugated ring structures, thereby, the weight loss was decreased. These findings demonstrated that OPFHA-MEA fibers had better thermal stability due to the higher degree of cyclization.

TG curves of the OPF and OPF via chemical pre-treatment.
Flame retardant property
The LOI of the pre-oxidized fibers was tested and the results were listed in Table 2. After pre-oxidation, the PAN fibers underwent cyclization, dehydrogenation, oxidation reactions, and so on. These reactions would convert the original linear macromolecular structures into heat-resistant planar trapezoidal structures and thus increased the LOI values of OPF. The OPF obtained from the pristine PAN fibers pre-treated with chemical reagents had a higher LOI value than the OPF obtained from the pristine PAN fibers without chemical pre-treatment. This was because chemical pre-treatment caused chemical reactions of functional groups (e.g. C≡N and C=O groups) on the macromolecular chains to produce new groups. The presences of new groups improved thermal property of PAN fibers, decreased the initial pre-oxidation reaction temperature and accelerated the pre-oxidation reaction rate. These aspects contributed to higher LOI value of fibers. The LOI value of the OPFHA-MEA reached 40.1, which was much higher than that of other OPF (i.e. OPFHA, OPFMEA, and OPFMEA-HA). On one hand, compared with individual chemical pre-treatment, the pre-treatment combined with HA and MEA caused more cyano groups on PAN macromolecular chains to be converted into amide groups, which was favorable for the formations of high-temperature resistant trapezoidal structures. On the other hand, it could avoid the problem of decreased conversion rates, which was caused by the groups’ spatial hindrances introduced to PAN macromolecular chains via pre-treatment in the order of MEA and HA.
Microscale combustion calorimetry (MCC) was also employed to measure the combustion property of OPF, and the relevant parameters including the peak of heat release rate (PHRR) and total heat released (THR), which were important indicators to reflect combustion characteristics and evaluate fire safety.30,31 In Figure 6(a), it was obvious that the pristine PAN fibers was flammable with a high PHRR of 303.4 W/g. The PHRR values of the OPF (93.8 W/g), OPFHA (69.5 W/g), OPFMEA (55.7 W/g), and OPFMEA-HA (55.6 W/g) were remarkably lower than that of pristine PAN fibers, and the PHRR of OPFHA-MEA exhibited a minimum value of 29.6 W/g among all the samples. The THR of all samples showed a similar downward trend with PHRR, as shown in Figure 6(b). The THR of pristine PAN fibers was 68.3 kJ/g and the THR of OPFHA-MEA was decreased to 8.0 kJ/g, which had ~88.3% reduction in comparison with PAN fibers. In comparison with PAN fibers, the THR values of OPF, OPFHA, OPFMEA, and OPFMEA-HA were 30.7, 20.2, 14.1, and 10.2 kJ/g, and were decreased by 55.1%, 70.4%, 79.4%, and 85.1%, respectively. Meanwhile, the OPFHA-MEA demonstrated good flame retardant performance with lowest PHRR and THR values, due to more amino groups reacted with cyano and ester groups on PAN macromolecular chains. This was favorable for the formations of cyclic pyridine structures with high temperature resistant.

Heat release rate (a) and total heat release and (b) curves of pristine PAN fibers, OPF, and OPF via chemical pre-treatment.
Conclusion
In this work, the techniques of chemical pre-treatment and pre-oxidation were used to improve the thermal stability, flame retardant and mechanical properties of OPF. The results showed that changes of characteristic absorption peaks with decreased cyano groups, disappeared ester groups, and presented amidoxime groups confirmed chemical reactions during pre-treatment on PAN fibers. The crystalline structure of the fibers happened to change due to the occurrences of cyclization reactions. The OPF had relatively smooth fibers’ surface with little grooves and cracks. The OPF were made from the pristine PAN fibers through chemical pre-treatment followed by pre-oxidation in air demonstrated excellent thermal stability, flame retardant, and mechanical properties, which attributed to the formed cyclic pyridine structures with high temperature resistant.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors acknowledge financial supports by Equipment Pre-research Joint Fund of Ministry of Education (6141A02022267), Jiangsu Universities “Qing Lan” Project ([2016] 15), Fundamental Research Funds for the Central Universities (JUSRP51621A, JUSRP41910), the Open Project Program of Jiangsu Advanced Textile Engineering Technology Center (XJFZ/2018/04), and the Open Project Program of Fujian Key Laboratory of Novel Functional Textile Fibers and Materials at Minjiang University (FKLTFM1801).