
Abstract
Background
Cognitive impairments commonly occur after traumatic brain injury (TBI) and affect daily functioning. Cortisol levels, which are elevated during acute hospitalization for most individuals after severe TBI, can influence cognition, but this association has not been studied previously in TBI.
Objective
We hypothesized that serum and cerebral spinal fluid (CSF) cortisol trajectories over days 0–5 post-injury are associated with cognition 6-month post-injury.
Methods
We examined 94 participants with severe TBI, collected acute serum and/or CSF samples over days 0–5 post-injury, and compared cortisol levels to those in 17 healthy controls. N = 88 participants had serum, and n = 84 had CSF samples available for cortisol measurement and had neuropsychological testing 6 months post-injury. Group based trajectory analysis (TRAJ) was used to generate temporal serum and CSF cortisol profiles which were examined for associations with neuropsychological performance. We used linear regression to examine relationships between cortisol TRAJ groups and both overall and domain-specific cognition.
Results
TRAJ analysis identified a high group and a decliner group for serum and a high group and low group for CSF cortisol. Multivariable analysis showed serum cortisol TRAJ group was associated with overall cognitive composites scores (P = .024) and with executive function (P = .039) and verbal fluency (P = .029) domain scores. CSF cortisol TRAJ group was associated with overall cognitive composite scores (P = .021) and domain scores for executive function (P = .041), verbal fluency (P = .031), and attention (P = .034).
Conclusions
High acute cortisol trajectories are associated with poorer cognition 6 months post-TBI.
Introduction
Traumatic brain injury (TBI) is a leading cause of disability and death globally. 1 Effective neuroprotective treatments are lacking due, in part, to heterogeneity in demographic and clinical characteristics and variability in acute secondary injury cascades post-injury.2,3 To inform prognostication and identify neuroreparative treatments, we previously examined inflammatory markers, sex hormones, and monoaminergic and neurotrophic molecules as potential biomarkers indicative of disability and mortality after TBI.2,4-7 Impaired cognition is one of the most common problems after TBI, occurring in ∼70% of individuals with severe injury. 8 Cognitive deficits broadly affect functioning, disrupting daily life activities, employment, social interaction, and life satisfaction.8,9 Early biomarkers specific to poor cognitive prognosis may inform outcome prediction and personalized cognitive dysfunction treatment approaches.
Neuroendocrine hormone profiles may be sensitive predictive markers for cognitive deficits after TBI. Acute TBI affects the hypothalamic–pituitary–adrenal (HPA) axis, which regulates the stress response and can mediate negative effects of inflammation on the central nervous system (CNS).2,6,10,11 In response to a stressor, the hypothalamus releases corticotropin-releasing hormone (CRH) which binds to receptors in the anterior pituitary gland, in turn releasing adrenocorticotropic hormone (ACTH). 12 The adrenal glands then produce cortisol in response to this ACTH signaling. We previously documented elevated cerebrospinal fluid (CSF) and serum cortisol profiles occurring over the first week after severe TBI6,10; elevated CSF cortisol was associated with worse 6- and 12-month global and disability outcomes. 10 Elevations can occur from disrupted negative feedback involving the HPA axis and either decreased glucocorticoid receptor sensitivity of HPA axis organs or decreased CRH, both of which may result in sustained elevated cortisol levels.13-15
Though cortisol can have transient beneficial anti-inflammatory effects, aiding in immune cell removal and inhibiting immune cell infiltration at injury sites in the periphery and CNS,11,16 elevated cortisol levels can lead to excessive or maladaptive inflammatory responses post-injury, 14 perpetuating blood brain barrier (BBB) 17 and neural cell dysfunction. 11 We have shown after severe TBI that elevated acute (days 0–5) CSF cortisol mediates the relationship between inflammatory cytokine profiles and 6-month post-injury outcome. 2 We have also established that CSF BDNF levels and their associated relationships with mortality differ based on CSF cortisol trajectories after TBI. 18 Serum cortisol levels reflect HPA axis response to TBI and associated injuries, since the adrenal glands are the primary contributors to systemic cortisol levels post-injury.6,10,19 Systemic cortisol transport into the CNS is the primary source of brain exposure to cortisol. This transport is highly regulated, but cortisol can also move passively into the CNS in the setting of BBB compromise.17,20,21 Our work shows CSF cortisol levels after TBI can be up to 10X that of healthy controls, and higher CSF cortisol levels measured from samples collected during the first week after injury are associated with mortality and survivor-specific functional outcomes after severe TBI.10,18 However, no one has explored how acute cortisol profiles affect cognition among TBI survivors. Our objective was to identify relationships between days 0–5 post-injury serum and CSF cortisol profiles and 6-month neuropsychological test scores after severe TBI. We hypothesized that consistently high serum and CSF cortisol profiles are associated with poor cognition measured 6 months post-TBI.
Methods
Recruitment
A cohort (N = 94) of adults with severe TBI (Glasgow Coma Scale (GCS) ≤8, with positive CT scan findings of TBI (per radiologist clinical report, not including isolated skull fracture)) was recruited from the University of Pittsburgh Medical Center as a part of an Institutional Review Board–approved study involving prospective biosample collection and outcome assessments. A schematic flow chart of how this study’s cohort was derived is shown (Figure 1). A healthy control cohort (n = 17) provided demographic information and serum and/or CSF samples, collected via lumbar puncture, for this study to generate reference values for cortisol measurements. For study inclusion, participants with TBI needed 1) at least two acute (day 0-5 post-injury) serum and/or two acute CSF cortisol measurements collected on different days during the day 0-5 sampling period, and 2) neuropsychological test data in at least one cognitive domain at 6 months post-injury. Consort-styled diagram of how the cohorts were derived. Of the 94 participants, 88 subjects had serum cortisol and at least one cognitive composite score and 84 subjects had CSF cortisol and at least one cognitive composite score. Due to variations in capacity for participants to complete neuropsychological testing, small differences in cognitive domain specific sample sizes occurred. The overlap between serum and/or CSF with cognitive composite scores resulted in a cohort of 94 people. N = 319 serum and N = 289 CSF daily cortisol values were analyzed (mean 3.6 serum values per individual; mean 3.4 CSF values per individual). The number of serum values for days 0–5 were 44, 60, 66, 59, 59, and 31, respectively. The number of CSF values for days 0-5 were 26, 61, 62, 59, 56, and 27, respectively. Note: TBI = traumatic brain injury, CSF = cerebrospinal fluid.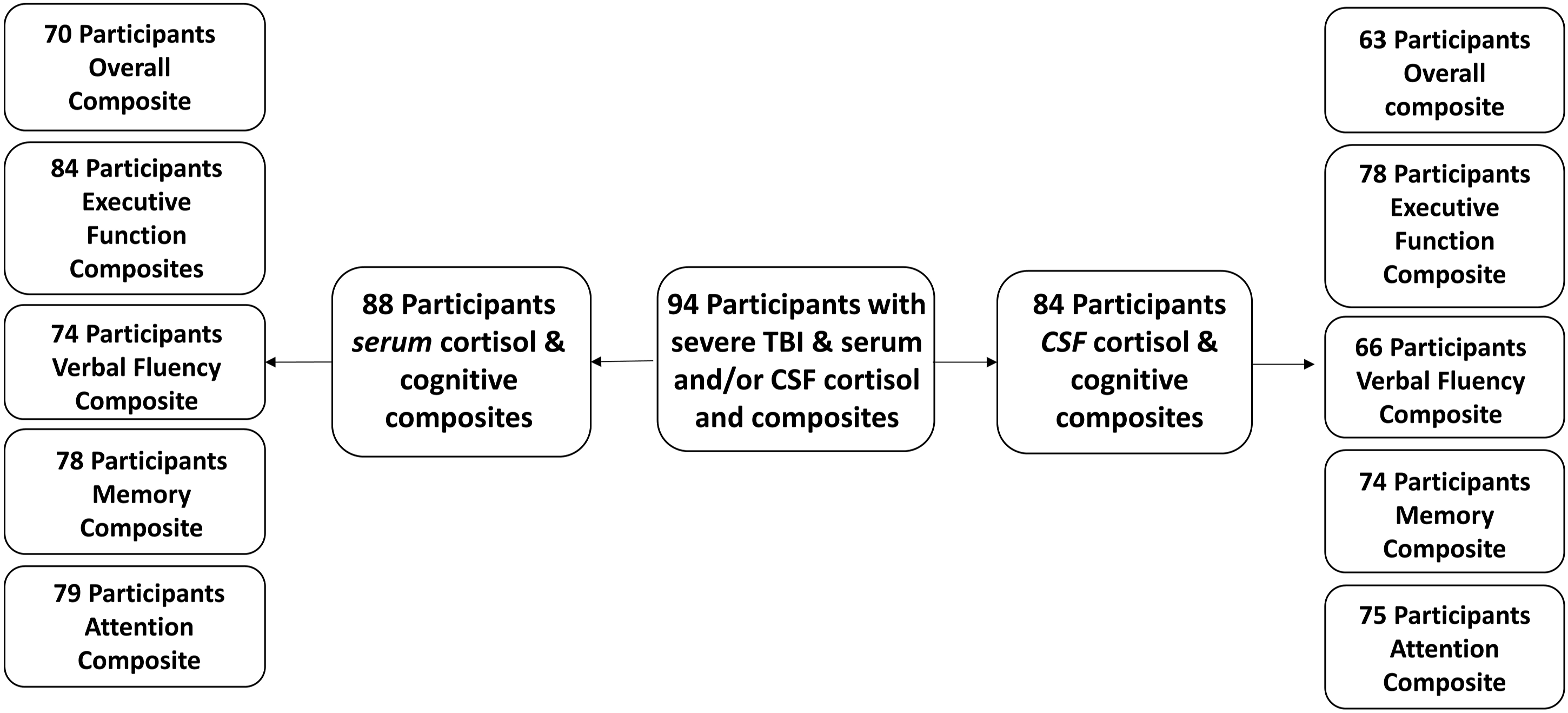
Of the 94 participants included in this study with serum and/or CSF cortisol levels and available cognitive composite scores, 88 had serum cortisol and 84 had CSF cortisol (Figure 1). Those who died prior to cognitive testing and TBI survivors unable to complete any cognitive testing were not included in this analysis.
Measures
Demographic and clinical variables
Demographic and clinical variables were abstracted from medical charts and self-reported through interviews. Given that severe TBI is accompanied by other extracranial injury and critical illness that may impact cortisol levels post-TBI, we collected information on extracerebral trauma and complications that ensued from these injuries. Abstracted variables included age, sex, race, hospital length of stay (LOS), and measures of trauma severity, including Injury Severity Score (ISS) and non-head ISS, mechanism of injury (MOI), injury complications, and the best GCS score within 24 hours of injury. Different injury types were extracted from head CT reports post-injury, including subdural hematoma, subarachnoid hemorrhage, diffuse axonal injury, epidural hematoma, contusion, intraventricular hemorrhage, intracerebral hemorrhage, and other findings.
Cognition
Cognition at 6 months post-injury was assessed via neuropsychological tests sensitive to cognitive deficits common after TBI. Cognitive composite scores were used to determine cognitive impairment based on eight neuropsychological tests assessing four cognitive domains described previously 22 (Supplementary Figure 1). The verbal fluency domain includes the Delis–Kaplan Executive Function Systems Verbal Fluency section 23 and the Controlled Oral Word Association Test. 24 The attention domain includes Trail Making Test A 25 and the Digit span subtest from the Wechsler Adult Intelligence Scale-R. 26 The memory domain includes The California Verbal Learning Test II-Long Delay Free Recall score 27 and the Rey–Osterrieth Complex Figure Test, 28 and the executive function domain includes the Trail Making Test B 25 and Stroop 29 Interference Score. Raw test scores were converted to norm-based t-scores, adjusting for age, race, sex, and education where applicable. If at least one test was completed in a domain, a domain-specific composite score (average of t-scores within the domain) was calculated. Overall cognitive composite scores were calculated as an average of the four domain-specific scores. A cognitive composite t-score of 50 is considered average for healthy individuals. Within each domain and for overall composite scores, cognitive impairment was defined as a t-score of ≤40.0 (1 SD below average).
Sample collection
Blood samples were collected in red top tubes every morning at ∼7:00
Not all TBI participants had samples collected every day. Sample missingness was attributable to lack of patient availability for blood/CSF sample collection, low CSF output, or EVD device removal prior to the conclusion of the day 0–5 sampling period. Cortisol data were binned into 24-hour intervals, and for bins with more than one sample per 24-hour time period (CSF samples), values were averaged for data analytic purposes, resulting in N = 319 serum and N = 289 CSF daily cortisol values across the day 0–5 period. Previous work suggests diurnal cortisol patterns are diminished shortly after moderate-to-severe TBI, as indicated by the fact that there were no significant differences observed in that study between morning and evening cortisol levels. 6
Cortisol measurements
Cortisol levels were measured for some serum and CSF samples using a solid phase 1251 radioimmunoassay (RIA) with a Coat-A-Count® In-vitro Diagnostic Test Kit (Siemens Health care Diagnostic Inc, Los Angeles, CA). The detection limit for this assay is 2 ng/mL. This kit is designed for direct cortisol measurement using 25 μL aliquots of sample. Inter- and intra-assay coefficients of variation (CV) were both less than 10% when using this methodology. The detection limit value was assigned to CSF and serum samples with out of range (low) values, while samples (n = 8 CSF samples only) were assigned a value of .001 ng/mL if levels were undetectable.
Cortisol levels for additional CSF and serum samples were measured using a commercial ELISA kit (1-3002, Salimetrics, PA, USA) according to manufacturer instructions. The detection limit for this assay is .07 ng/mL. To avoid matrix effects with this assay, serum and CSF samples were diluted 1:36 and 1:4, respectively. Although the kit was developed and validated for saliva, our pilot work (Supplementary Figure 2) using serum cortisol showed an excellent profile of linearity with serial dilution, and recovery (90%–110%) for standards with a range of dilution 1:20 to 1:40. Among the 96 wells in each plate, 10 wells were used in duplicate to evaluate intra-plate reliability, and six additional wells were used to evaluate inter-plate reliability. Otherwise, all samples were evaluated in singlet to conserve sample volume. The observed intra- and inter-plate variance were <5% CV and <6% CV for serum, and <10% CV and <16% CV for CSF, respectively.
Previous work using these assay platforms suggests a high degree of linearity between serum and saliva cortisol levels.
30
Notably, serum levels in this study were measured using the same RIA platform, and CSF samples were measured using the same ELISA kit noted in this study. Thus, linear regressions were generated by re-running samples to estimate RIA cortisol values for samples measured via ELISA to pool data for analysis. To accomplish this, we measured serum and CSF cortisol levels in a subset of samples (N = 38 serum; N = 30 CSF) for which we also previously measured cortisol using RIA in order to determine a correlation and generate a linear equation used to convert ELISA sample values and pool measurements across assay type to form one dataset. Serum cortisol levels for N = 38 samples run via both RIA and ELISA correlated were used to create the linear regression equation of
Statistical analysis
Statistical analyses were conducted using SAS (Version 9.4). Mean and standard error of the mean were calculated for age, hospital LOS, years of education, and Injury Severity Score (ISS), while median and interquartile range were determined for best GCS in 24 hours. Although an initial GCS score of ≤8 was required for study inclusion, the best GCS score in 24 hours was used in analysis as a better measure of injury severity that is less subject to bias from intubation or paralytics at initial time of injury, as previously described. 22 Frequencies and percentages were calculated for categorical variables. Demographic and clinical variables were compared between trajectory analysis (TRAJ) group membership and by cognitive impairment status, measured by overall composites scores, using Mann–Whitney U tests for continuous variables or χ2 or Fisher’s exact tests for categorical variables. Mann–Whitney U tests were used to assess relationships between TRAJ groups and cognitive impairment.
Trajectory analysis formulation
Group-based trajectory analysis (PROC TRAJ function in SAS) identified groups of individuals with similar longitudinal cortisol profiles using methods similar to previous work.2,5,10,18 Individuals needed to have at least two serum or two CSF samples, collected at any two distinct days post-injury, to be included in the respective serum TRAJ or CSF TRAJ analysis for longitudinal characterization. Both serum and CSF assay values underwent natural log-transformation prior to conducting TRAJ analysis. TRAJ group designations were based on a data driven, algorithmic procedure, rather than a clinical cut-point designating high cortisol levels. Bayesian Information Criteria and posterior probabilities were compared between models to determine optimal number of groups and polynomial order. Posterior probability represents the probability that, given the observed values an individual has, they belong to a given trajectory group, independent of the outcome data collected, and posterior probabilities are the basis for judging the adequacy of the model. For each individual in the cohort, the TRAJ group with the largest posterior probability is used to select group membership, and generally an average posterior probability ≥.7 for TRAJ groups generated for the cohort is considered acceptable. 5 Descriptive TRAJ group designations were provided for both CSF and serum that characterize relative differences in cortisol levels between groups.
Multivariable linear regressions assessed predictors of 6-month cognition using overall and domain-specific cognitive composite scores, adjusting for age, sex, and education as covariates.31-33 To further control for potential confounding with multivariable analysis, we included variables with significant associations with serum cortisol, CSF cortisol, and/or cognitive composite scores at the P < .1 level.
Results
Demographic, Clinical, and Cortisol Information
Demographic and clinical information for all participants with either serum and/or CSF cortisol and at least one composite score and demographics split by serum and CSF.
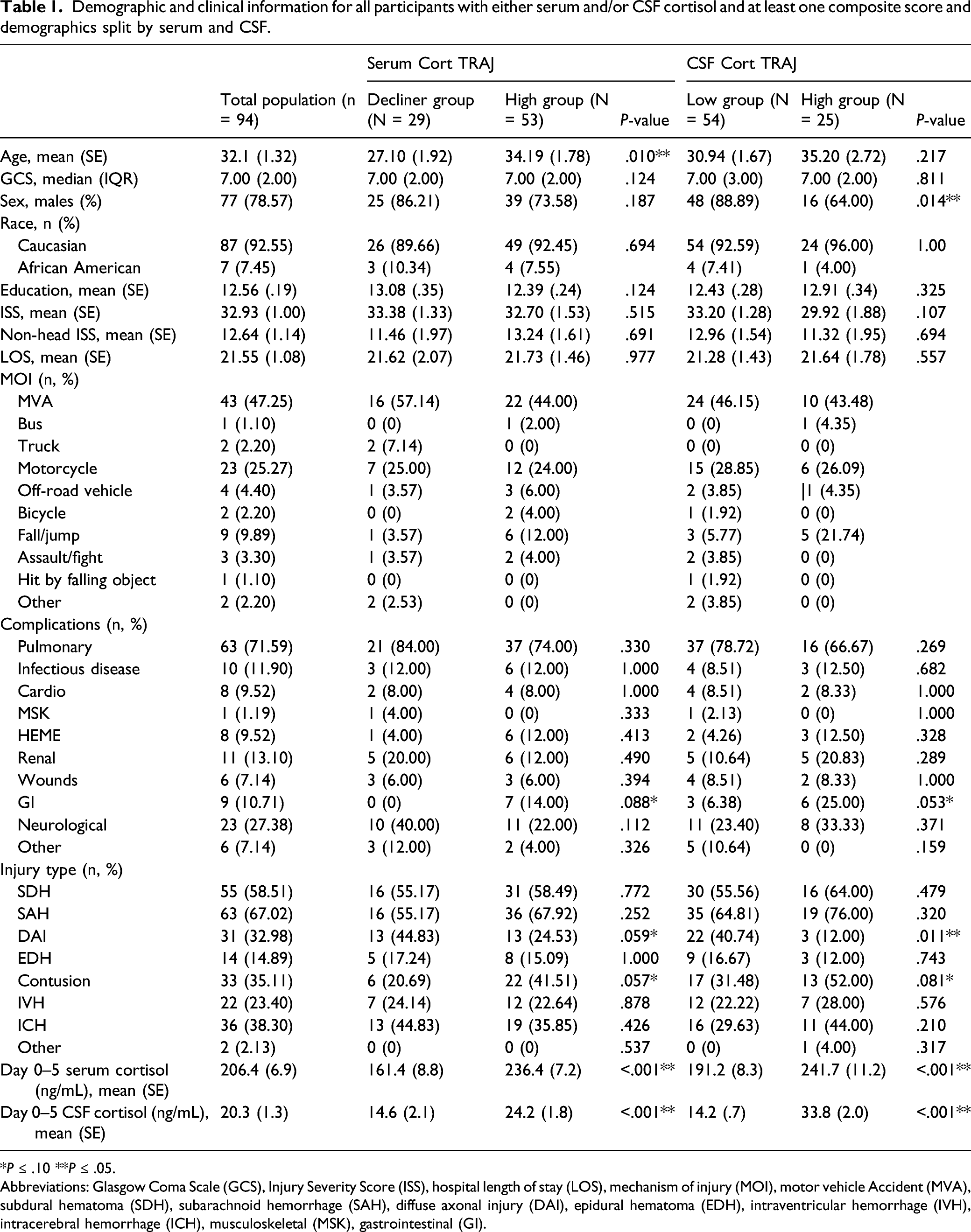
*P ≤ .10 **P ≤ .05.
Abbreviations: Glasgow Coma Scale (GCS), Injury Severity Score (ISS), hospital length of stay (LOS), mechanism of injury (MOI), motor vehicle Accident (MVA), subdural hematoma (SDH), subarachnoid hemorrhage (SAH), diffuse axonal injury (DAI), epidural hematoma (EDH), intraventricular hemorrhage (IVH), intracerebral hemorrhage (ICH), musculoskeletal (MSK), gastrointestinal (GI).
Serum and CSF Trajectories
For serum cortisol, TRAJ analysis identified a consistently high group and a decliner group. Average posterior probabilities were 90.51% and 79.39% for the high group and decliner group, respectively. The high group had significantly higher serum cortisol levels than controls on all days of testing, while the decliner group had serum cortisol levels similar to controls for the entire monitoring period. CSF cortisol TRAJ analyses identified a high group and low group. Individuals in both groups had higher CSF cortisol levels than control values over the entire monitoring period. Average posterior probabilities were 83.18% and 93.56% for high and low groups, respectively. Participants in the high serum cortisol group were significantly older than those in the decliner group (P = .010). There also was a greater proportion of men in the low vs high CSF cortisol group (P = .014). Diffuse axonal injury was more frequent in the low vs high CSF TRAJ groups (P = .011).
Factors Associated With 6-Month Cognition
Demographic and clinical characteristics associated with cognition.
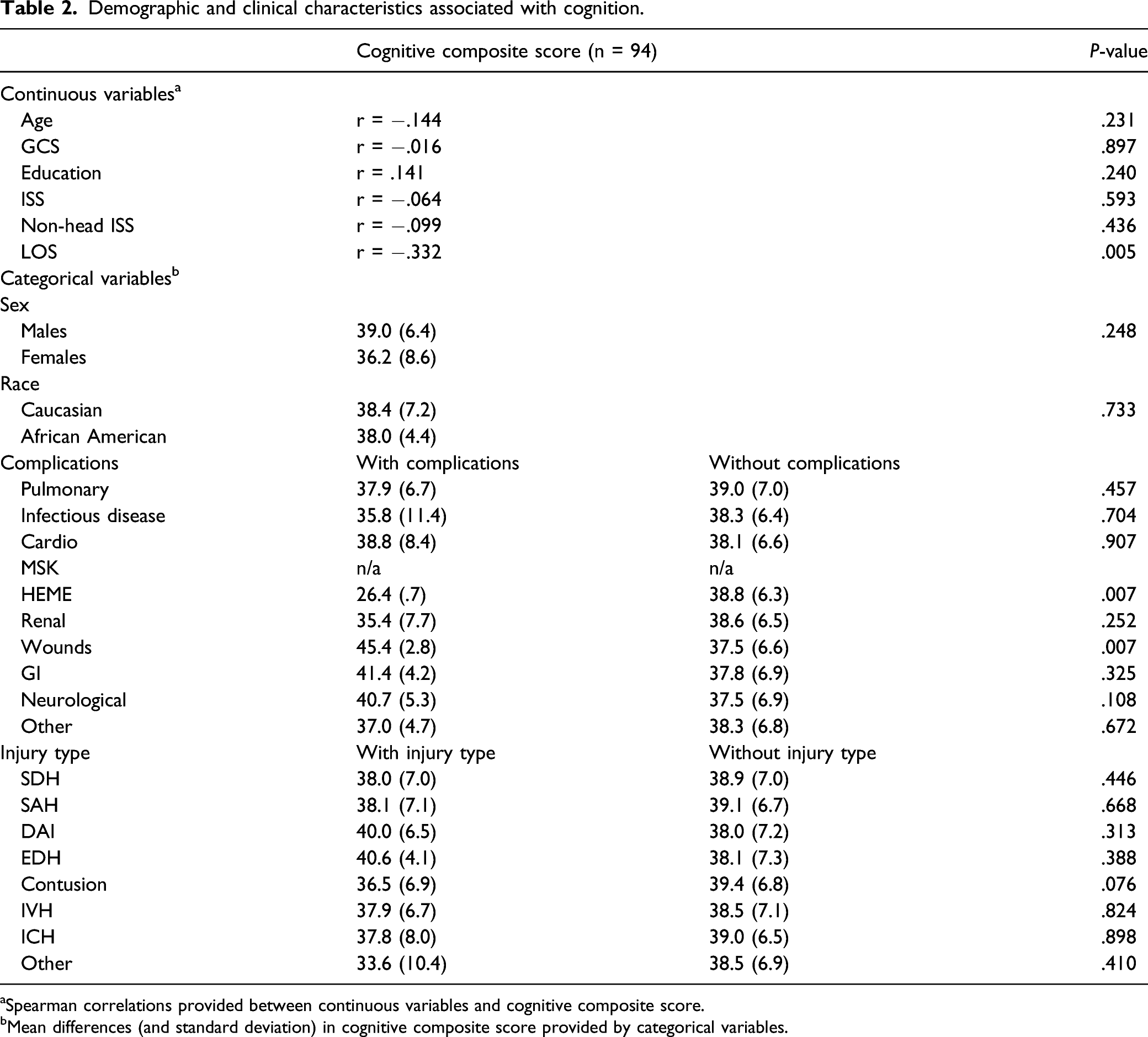
aSpearman correlations provided between continuous variables and cognitive composite score.
bMean differences (and standard deviation) in cognitive composite score provided by categorical variables.
Cortisol Associations With TRAJ Group Membership
Average daily serum and CSF levels were calculated by TRAJ group for each day (0–5) post-injury (A) Acute Serum Cortisol by TRAJ Group (N = 82). Daily average serum cortisol levels (ng/mL) were determined within each TRAJ group, and the error bars represent the standard error of the mean values for each day. Average serum cortisol levels were significantly different by TRAJ group for days 1–4 (P ≤ .05*), and there was a trend for differences in cortisol levels on day 5 (P = .0519‡). Control group mean level for serum cortisol was 139.8 ng/mL (SE: 11.9, SD: 48.9). (B) Acute CSF Cortisol by TRAJ Group (N = 79). Average CSF cortisol levels (ng/mL) were determined by day within each TRAJ group, and the error bars represent the standard error of the mean values for each day. Average CSF cortisol levels were significantly different for days 0–5 (P ≤ .05*) between the two TRAJ groups. Control group mean level for CSF cortisol was 4.5 ng/mL (SE: .4, SD: 1.5). Note: TRAJ = trajectory analysis, CSF = cerebrospinal fluid.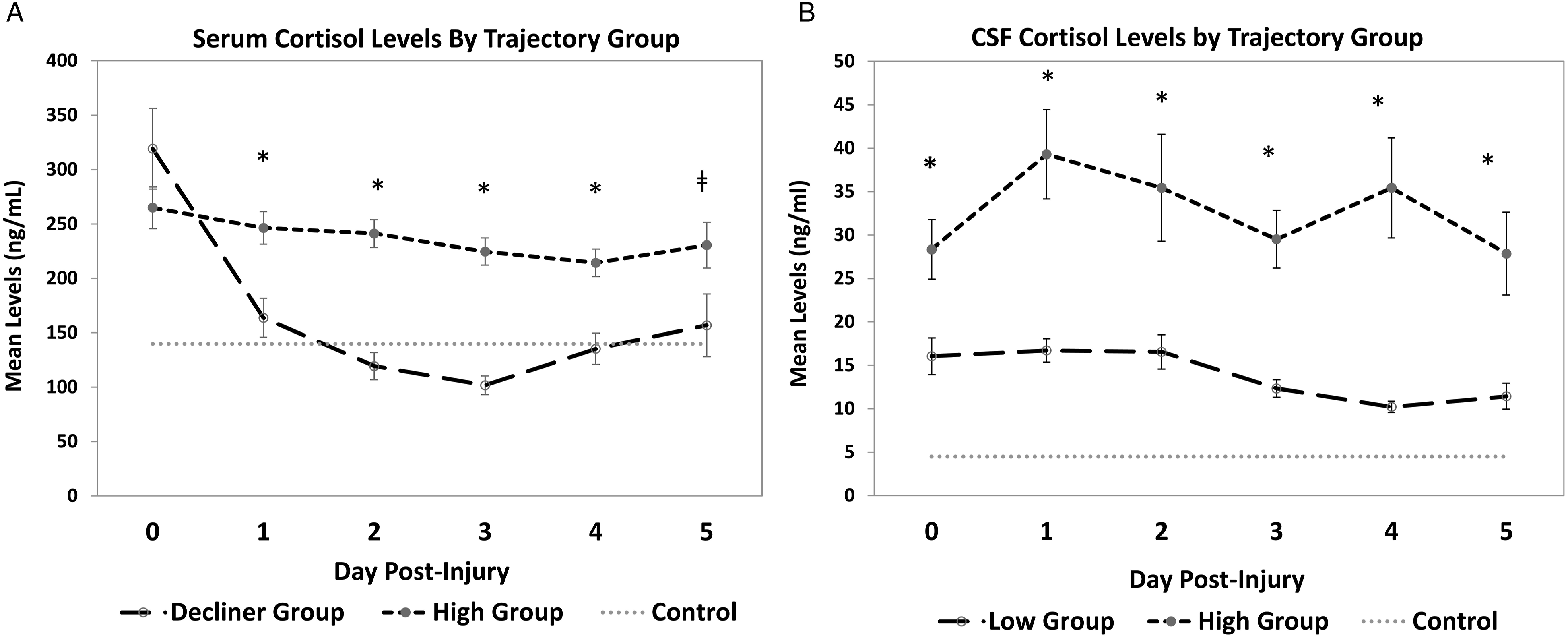
Trajectory Group Membership Associations With Cognitive Composite and Domain Scores
Average Composite Scores by Serum and CSF TRAJ Groups.
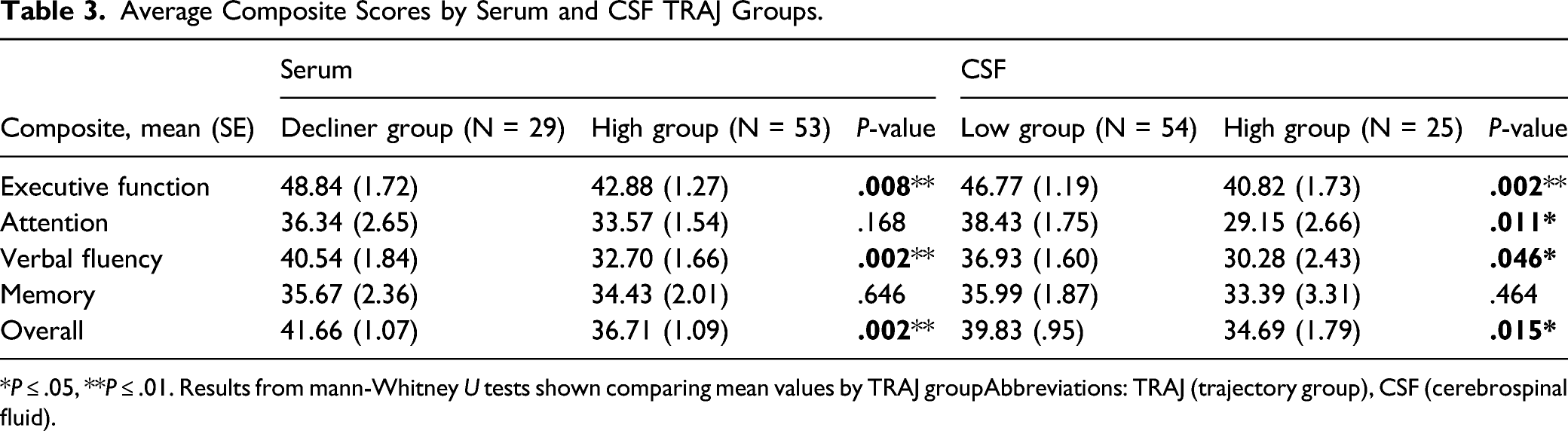
*P ≤ .05, **P ≤ .01. Results from mann-Whitney U tests shown comparing mean values by TRAJ groupAbbreviations: TRAJ (trajectory group), CSF (cerebrospinal fluid).
Serum Cortisol TRAJ is Associated With 6-Month Cognition
Multivariable linear regression of overall composites and domain scores at 6 months For serum and CSF cohort.
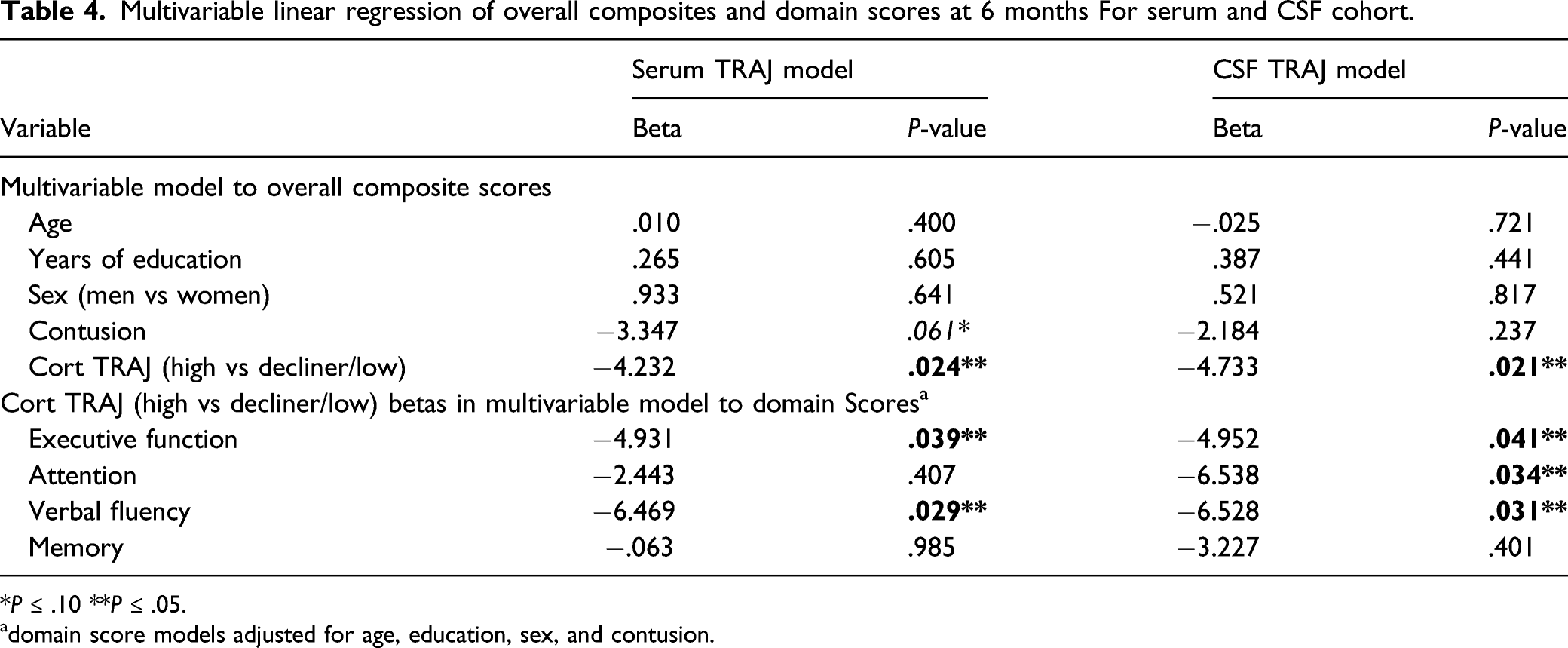
*P ≤ .10 **P ≤ .05.
adomain score models adjusted for age, education, sex, and contusion.
CSF Cortisol TRAJ is Associated With 6-Month Cognition
Similar models were generated for CSF TRAJ groups (Table 4). Adjusting for age, sex, GCS, years of education, and contusion, there was a 4.733 point lower overall composite scores in the high CSF group compared to the low group (P = .021). Executive function [−4.952 points; (P = .041)], attention [-6.538 points (P = .034)], and verbal fluency (–6.528 points (P = .031)) were also lower in the high vs low CSF TRAJ group. Memory, however, did not differ by CSF TRAJ group (P = .401).
Discussion
The novel association we found between acute cortisol profiles and later cognitive impairment after severe TBI suggests cortisol may be a potential early treatment target to improve long-term cognition post-TBI. Our results indicate those with acute serum and CSF cortisol profiles that are high and remain high over days 0–5 post-injury have poorer cognition at 6-months post-injury than those whose cortisol profiles are consistently low or may start high but decline early post-injury. This finding suggests acute cortisol trajectories among TBI survivors are an early indicator of ongoing hypercortisolemia, which subsequently affects later cognition. If these results are confirmed, early cortisol trajectories could be used to identify “at-risk” individuals who may benefit from therapies that modulate cortisol production and signaling.
In the absence of TBI, HPA axis feedback loops associated with elevated serum cortisol levels signal to the pituitary and hypothalamus to decrease ACTH and CRH production, thus decreasing further cortisol production. However, multiple cytokines (e.g., IL-6, TNFα) can contribute to sustained cortisol elevations 34 such as what occurs in response to trauma,2,13 but cortisol elevations can also result from reduced glucocorticoid-mediated feedback to the HPA axis. 15 This latter phenomenon has been examined in rats, where HPA feedback may be disrupted by changes in glucocorticoid receptor expression. 35 Although cortisol has been extensively studied, little evidence exists regarding mechanistic cortisol effects peripherally or centrally after TBI. Cortisol production occurs primarily in the adrenal glands, 19 although some CNS-derived hormone synthesis may occur. 19 Further, excess serum cortisol may enter the CNS after TBI due to P-glycoprotein dysfunction, an important BBB transporter that limits accumulation of drugs and hormones like cortisol in the brain.17,20,21 Our findings align with this research, in that those with higher serum and CSF cortisol profiles had worse cognition.
Relevant to the acute stress response associated with TBI, differential cortisol receptor binding may contribute to how stress-induced elevated cortisol levels affect cognition. 36 Cortisol binds with both mineralcorticoid (MR) and glucocorticoid receptors (GR). 37 MR’s have a higher affinity for cortisol and are the main receptors maintaining cortisol mediated circadian rhythm. 37 Cortisol receptors are widely distributed in the CNS, wherein MR’s are present in the limbic system and GR’s are present in both subcortical and cortical regions, and affect cognition. 37 Differential receptor occupation during stress negatively affects hippocampal function. During stress-induced hypercortisolemia, similar to accumulation of cortisol acutely after TBI, MR’s become saturated, resulting in increased GR binding. 37 Experimental TBI work in fluid percussion has demonstrated aberrant stress responses after injury,13,38 yet the work linking dysfunctional stress responses after TBI is both complex and likely temporally and receptor dependent. Previous experimental work suggests GR inhibition with mifepristone can mitigate a disrupted neuroendocrine stress response. 39 Other experimental studies suggest MR stimulation, in the setting of reduced plasma corticosterone, improves spatial memory early after TBI 40 and that GR down-regulation and MR upregulation are both necessary in the setting of low endogenous corticosterone levels to promote neuronal survival and spatial learning and memory early after TBI. 41 Future work is needed to elucidate how cortisol stress responses are disrupted clinically after TBI and how CNS GR/MR balance during TBI recovery affects learning and memory over time. Future work may also require specific focus on age and sex HPA response moderators,42,43 in appropriately sized TBI and control cohorts, particularly given some indication in our survivor data that age and sex affect early cortisol profiles post-TBI.
Imaging studies characterizing CNS cortisol effects on specific brain structures suggest structure-specific vulnerability to elevated cortisol levels. One study using pharmacological functional magnetic resonance imaging found that acute hydrocortisone administration resulted in a time dependent increase in hippocampus blood-oxygen level–dependent signals, indicating cortisol may have rapid effects on structures like the hippocampus. 44 Another study reported that long term effects of excessive cortisol exposure due to Cushing’s syndrome led to brain volume loss and grey and white matter abnormalities that affect the hippocampus and medial frontal and anterior cingulate gyri. 45 These changes may be reversible after cortisol level correction.45,46 Given knowledge of ongoing brain volume atrophy in the context of TBI 31 and our findings of acute hypercortisolemia, future work should evaluate if acute cortisol exposure contributes to or accelerates TBI-related brain atrophy. The role of persistent hypocortisolemia on brain atrophy post-TBI should also be explored. Prior reports using a larger cohort from our center suggest that acute adrenal insufficiency (AI) can occur after severe TBI, but AI status did not adversely affect acute care mortality, vasopressor or corticosteroid use, or 6 month global outcome. 6
Although our work only evaluates cortisol levels over days 0–5 post-injury, our data suggest high cortisol levels early after TBI may portend ongoing hypercortisolemia that contributes to long-term cognitive impairments. If true, hypercortisolemia may affect entire brain network function after TBI. 47 More research is needed to examine how cortisol affects cognitive neural networks, and associated vulnerable structures, after TBI. For example, elevated levels may persist and lead to ongoing increases in GR binding involving brain regions relevant to cognition. Differential binding affinities associated with MR’s vs GR’s may leave regions with higher GR densities relative to MR’s vulnerable to damage due to hypercortisolemia. 48 Further, adverse effects of hypercortisolemia on plasticity and neurogenesis mechanisms after TBI may result from specific receptor activation. For example, MR agonists can increase long-term potentiation (LTP) while GR activation can decrease LTP. 49 TBI recovery relies on injury-induced increases in plastic activity to support brain repair, which we hypothesize may be hampered in the setting of hypercortisolemia. 50
We note that trajectory groups in both serum and CSF have cognitive composite scores below normal (<50), and we describe domain-specific and total cognitive composite scores at levels considered cognitively impaired (<40). However, the lower scores in the high trajectory groups suggest a cortisol dose-response effect. Clinically meaningful differences in cognitive testing may generally be detected at .5 standard deviation differences, 51 representing a 5-point difference in the current study, which we observed between trajectory groups in several domains. Memory domain performance in our study was impaired in both high and low trajectory groups but were not different between groups in serum or CSF, suggesting there may be a ceiling effect for cortisol with respect to memory relative to other domains. Memory impairment also is fairly nonselective and universal after moderate-to-severe TBI, 52 potentially explaining why cortisol may have less specific direct effects on memory compared to other domains. Other reasons for memory being insensitive to acute cortisol profiles may include the neuropsychological tests selected for this study, sample size, and the exclusion of those too severely injured to complete testing.
Higher education and hospital LOS were also associated with cognitive performance. Higher education has been associated with more cognitive reserve and better outcomes after TBI, 53 and longer LOS has been associated with more severe injuries and with older age, 54 both of which are consistent with our findings. Contusion was marginally associated with both CSF and serum cortisol levels and also with cognitive composite scores. Although our review of the literature finds no evidence that contusions increase cortisol levels, one might surmise that the intraparenchymal blood and lesion burden associated with contusion might affect cortisol levels. However, multivariable analyses show that CSF and serum cortisol TRAJ remained significantly associated with cognitive composite scores, even when adjusting for contusion. Thus, despite the postulation that intraparenchymal blood associated with contusions may affect CSF and serum cortisol, our data show that cortisol independently affects cognition.
This study has several limitations and future directions to consider. Our cohort was limited to survivors with acute cortisol levels and 6-month cognitive testing. Individuals cognitively unable to complete neuropsychological testing were excluded, so cognition in this cohort may not accurately represent more severe cognitive dysfunction. Since this study only examines acute cortisol profiles in patients, we cannot conclude if early elevated cortisol directly causes these cognitive deficits or simply reflects the start of a chronic hypercortisolemic state that contributes to cognitive impairment. However, rates of chronic central hypoadrenalism range from 5% to 46% after TBI, 55 suggesting that hypercortisolemia is likely to be a time-limited phenomenon and that acute cortisol exposure may contribute significantly to cognition, in its own right, after TBI. Yet, it remains unknown how acute cortisol profiles correspond to cortisol measurements in subsequent months post-injury, 55 particularly in the setting of ongoing inflammation post-TBI. 56 Unmeasured health and environmental post-acute factors may also influence stress and cortisol levels, including socioeconomic challenges, caregiver support, or other adverse life or health events occurring between initial hospitalization due to TBI and 6-month outcomes. As an observational study, we also cannot determine if cortisol profiles directly influence cognition or are indicators of other processes affecting cognition. We assessed serum/CSF cortisol levels, but hair or urinary cortisol may offer more stable measurements that reflect total cortisol exposure over time, particularly when monitoring TBI recovery. Salivary cortisol profiles post-TBI may be feasible in tracking diurnal cortisol variation for those cognitively able to complete this type of complex testing in home and community settings.
Cortisol secretion is not the only stress response factor that may affect cognition. For example, catecholamines can be affected by stress, 57 and alterations may perpetuate cognitive decline in the setting of TBI.7,32 Since inflammatory signaling regulates both HPA and catecholamine pathways, inflammatory pathway relationships to both catecholamine and cortisol production over time should be assessed together for their independent and interrelated impacts on cognitive function after TBI. Hypothalamic–pituitary–adrenal axis function, inflammation, and catecholaminergic tone can all be modulated by lifestyle interventions like yoga 58 and mindfulness, 59 presenting therapeutic non-pharmacologic, rehabilitation-relevant interventions for at-risk individuals that may improve cognitive outcomes after TBI.
Supplemental Material
sj-pdf-1-nnr-10.1177_15459683211048771 – Supplemental Material for Acute Cortisol Profile Associations With Cognitive Impairment After Severe Traumatic Brain Injury
Supplemental Material, sj-pdf-1-nnr-10.1177_15459683211048771 for Acute Cortisol Profile Associations With Cognitive Impairment After Severe Traumatic Brain Injury by David J. Barton, Raj G. Kumar, Alexandria A. Schuster, Shannon B. Juengst, Byung-Mo Oh and Amy K. Wagner in Neurorehabilitation and Neural Repair
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National Institute on Disability and Rehabilitation Research (Grant Number: NIDILRR 90DP0041, Wagner); US Department of Defense (Grant Number: W81XWH-07-1-0701, Wagner); National Institutes of Health (Grant Number: TL1TR001858, Kumar; Grant Number: T32HL134615, Barton); Centers for Disease Control (Grant Number: R49 CCR 323155).
Supplementary Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.