
Abstract
Spinal cord injury (SCI) is a devastating event that often leads to permanent neurological deficits. Evidence from emerging studies has implicated oxygen-derived free radicals and high-energy oxidants as mediators of secondary SCI. Therefore, targeting these mediators using antioxidants could be beneficial for the disease. Several signaling pathways, such as the nuclear factor erythroid-2-related factor 2/heme oxygenase 1 (Nrf2/HO-1), have been associated with the regulation of some pathophysiological features of SCI. Curcumin is a plant medicinal agent whose diverse pharmacological properties have been extensively investigated and reported, notably its ability to curtail inflammatory damage by inhibiting the nuclear factor-κ-light-chain-enhancer of activated B cells. In this review, we analyze the role of curcumin in activating Nrf2/HO-1 and scavenging free radicals to repair SCI. With its minimal side effects, curcumin could be a potential therapy for SCI treatment.
Spinal cord injury (SCI) is a distressing condition with high morbidity and mortality.1,2 SCI can have both physical and psychological effects on the individual, family, and society. 3 At present, there are no effective clinical treatments for SCI, partly because of the complexity of its pathophysiological process. Thus, understanding its pathophysiological mechanism and developing an ideal, effective and viable repair strategy for neural regeneration and functional recovery after SCI are paramount.
The failure of SCI repair could be attributed to both the extrinsic inhibitory environment and a lack of intrinsic neuronal regeneration. 4 Particularly, the main factors that directly affect neural regeneration and repair following SCI are the damaged areas, 5 such as local ischemia, vascular network loss, disruption of the blood–spinal cord barrier, continuous release of inflammatory mediators, and formation of reactive astroglia scars, 6 which subsequently leads to degeneration of neurons, disintegration of fibers, and collapse of growth cones and further impedes axonal regeneration.7-9
Although there are no effective therapies for SCI currently, signaling pathways might play an important role in SCI repair. 10 For instance, nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the Cap’n’Collar basic leucine zipper (CNC-bZIP) transcription factor family, is an intracellular redox-sensitive switch. Once cells are exposed to either oxidative stress or chemopreventive factors, Nrf2 is dissociated from Keleh-like ECH-associated protein (Keap1), is translocated into the nucleus, and regulates the transcription of antioxidant genes.11,12 Activation of Nrf2 in adult rat’s neurons and astrocytes could improve spinal cord ischemia-reperfusion injury, because in Nrf2 knockout mice, neurological deficits in spinal cord tissues were exacerbated after SCI surgery.13,14 Hence, one possible approach of controlling oxidative stress and hindering apoptotic cascades could lie in the activation of the Nrf2 signaling in damaged spinal cord tissues.
Nrf2/Heme Oxygenase 1 (HO-1) Signaling Pathway and SCI
The Constituents of the Nrf2/HO-1 Signaling Pathway
Nrf2 belongs to CNC-bZIP member of the transcription activator family, including 7 domains, Neh1-7; the negative regulator Keap1 contains 5 domains, which are the N-terminal region, broad-complex tram track and bric-a-brac (BTB), intervene region (IVR), Di-glycine repetition region (DGR), and C-terminal region. 15 In the physiological state, the Neh2 domain of Nrf2 and DGR junction of its negative regulator Keap1 postsynaptic sites are located in the cytoplasm in the Keap1 functional domains, BTB and IVR. These regions are subjected to Cul3/Rbx1 E3 ubiquitination degradation that maintains a stable concentration.16,17
Under stress conditions, such as reactive oxygen species (ROS) and electrophilic groups, Keap1 conformational change of SH-1 group and Nrf2 phosphorylation cause uncoupling, with Nrf2 released into the nucleus. Nrf2 regulates the activities of target genes, such as superoxide dismutase (SOD), catalase (CAT), and phase II detoxification enzyme to remove ROS and other harmful substances after Nrf2 forms a heterodimer with Maf protein through Neh1 domain and combines with antioxidant reaction element (ARE). 13
HO-1 is one of the phase II detoxification enzymes and the subordinate signaling pathway triggered by HO-1 that protects multiple organs from oxidative stress. Combined with nicotinamide adenine dinucleotide phosphate (NADPH) and cytochrome P450, HO-1 catalyzes the cleavage of heme into Fe2+, carbon monoxide, and biliverdin, consequently converting to bilirubin through biliverdin reductase, and forms endogenous protective substances to regulate cell oxidation, apoptosis, and other biological cell activities. 18
The Nrf2/HO-1 signaling pathway is involved in multiple activities, including antioxidation, anti-inflammation, reduction of mitochondrial damage, and regulation of cell death, all these which can affect the outcome of a disease (Figure 1).
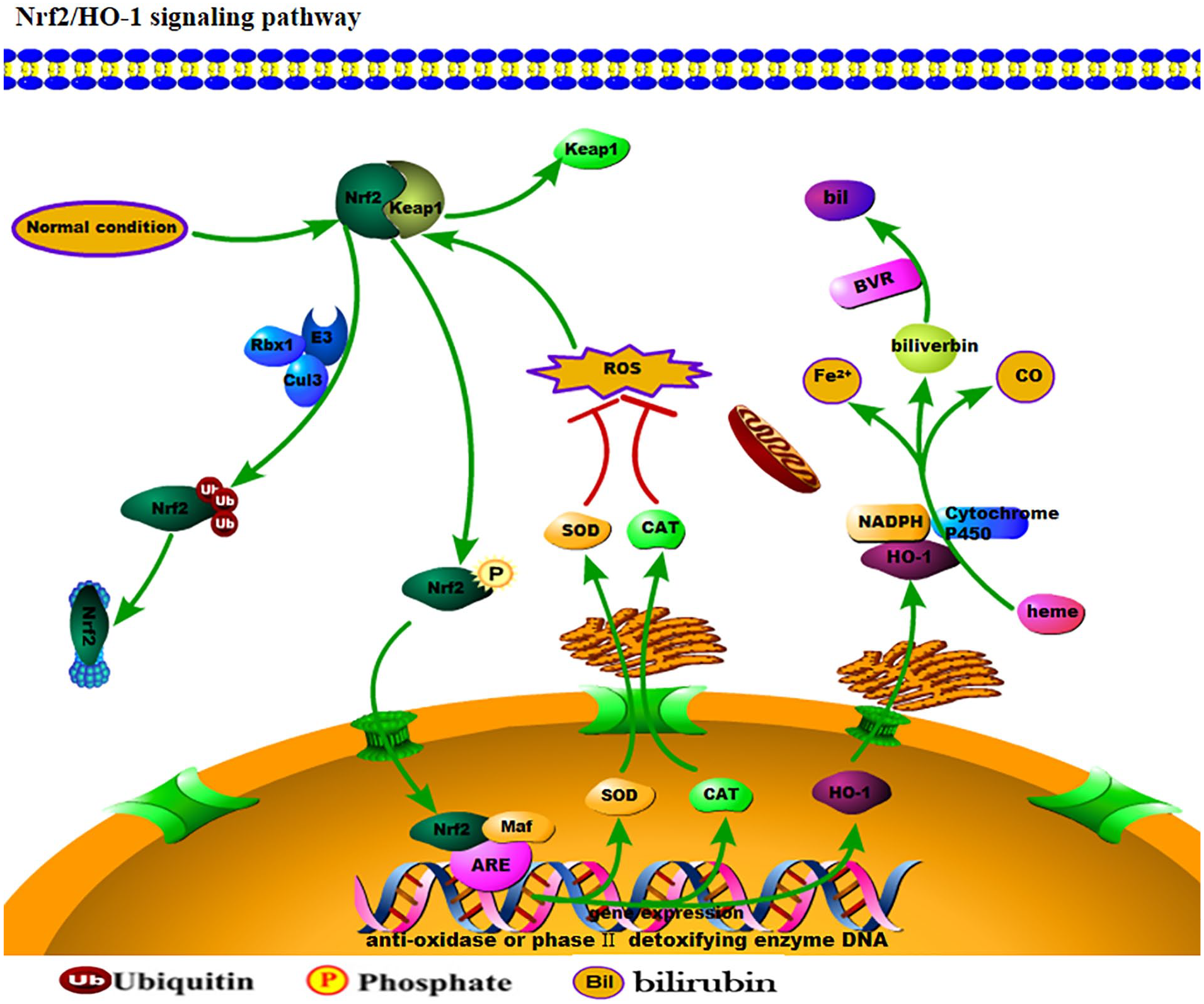
The Nrf2/HO-1 signaling pathway. Under normal conditions, Nrf2 in the cytoplasm is recruited by the Keap1 molecule, and hydrolyzed by the protease system under the actions of Rbx1, cul3, and E3 ubiquitin. During oxidative stress, Nrf2 in the cytoplasm is phosphorylated and isolated from Keap1 and binds to Maf after entering the nucleus. Thereafter, an ARE protein upregulates the expressions of SOD, CAT, and HO-1. SOD and CAT enter the cytoplasm and play a direct role in antioxidation. HO-1 degrades Heme into bilirubin under the action of NADH and P450, in response to oxidative stress.
The Biological Function of the Nrf2/HO-1 Signaling Pathway
The Nrf2/HO-1 signaling pathway plays a key role in inflammation and oxidative stress. In treating myocardial ischemia-reperfusion injured rats with resveratrol, the Nrf2/HO-1 signaling pathway downregulated the level of myeloperoxidase and upregulated the antioxidant levels of SOD, glutathione peroxidase, and CAT. 17 Previous studies have found HO-1 activation to curtail both the remodeling of cytoskeletal actin and the migration of CXLL-related polymorph nuclear leukocytes to the alveolar compartment as well as reduce microvascular endothelial permeability and stabilize lung barrier function, ultimately exerting anti-inflammatory effects.19,20 Thus, nonmyelinated HO-1 could be a significant player in anti-inflammation and provide a new perspective on anti-inflammatory mechanisms. Sulforaphane (SFP) is an activator of Nrf2. In an advanced glycation end products–induced rat hippocampal oxidative stress injury model, SFP significantly reduced the levels of tumor necrosis factor (TNF)-α and interleukin (IL)-1 associated with cell death in hippocampal tissues while increasing the activities of SOD, Glutathione peroxidase (GPx), and CAT.5,21 Erythropoietin can reduce the pyrogenation of neurons induced by sevoflurane through upregulating Nrf2, HO-1, and SOD protein levels in the neuronal culture while downregulating malonaldehyde protein levels.22,23 Thus, the Nrf2/HO-1 signaling pathway could be a combination of antipyrogenation and anti–lipid peroxidation. The Nrf2/HO-1 signaling pathway can have protective effects on the spinal cord and brain via its anti-inflammatory and antioxidant capacities.
The Role of the Nrf2/HO-1 Signaling Pathway in SCI
Oxidative and nitrosative stress are involved in the progression of SCI. The pathophysiology of SCI manifests itself as primary injury immediately after trauma, and the materialization of the secondary injury comes afterward in the later stage, which leads to excessive ROS production. 24 Increased reactive oxygen levels can trigger a series of complex molecular and cellular processes, including the activation of oxidative stress and inflammatory-related signaling pathways. 25 Various severe stress and inflammatory reactions can last for hours, days, or weeks and lead to long-term neurological dysfunction. Therefore, reducing ROS production, concomitant with curtailing oxidative stress and inflammatory response, could be paramount in preventing and treating acute spinal cord trauma. Under pathological conditions, activated leukocyte and macrophages generate considerable amounts of superoxide anion (O2•−) and other ROS, including hydrogen peroxide and hydroxyl radical. NADPH oxidase, which was originally found to be the primary source of O2•− in phagocytes, has emerged as the main source of O2•− in various other tissues, including central nervous system tissue.11,26 Studies have demonstrated the knockdown of type 2 ryanodine receptor gene to downregulate NADPH oxidase 2 expression, reduce oxidative stress, improve mitochondrial dysfunction and endoplasmic reticulum stress, and inhibit inflammation, concomitant with improved SCI recovery.27,28
The primary oxygen free radical produced in the body is O2•−, a highly reactive molecule. Superoxide is usually produced in the course of cellular respiration by mitochondria and as a product of certain oxidase enzymes in the cytoplasm. Oxygen free radicals react with NO to produce highly reactive and cytotoxic products, such as peroxynitrite and peroxynitrous acid. 29 Uncontrolled oxygen free radicals and peroxynitrite can cause cytotoxicity by damaging proteins, nucleic acids, and lipids and attack the double bonds of unsaturated fatty acids to cause lipid peroxidation.12,30 This radical lipid-free interaction produces peroxides, which can increase membrane damage because of its specific reactivity. In particular, neuronal tissues, such as the spinal cord, are highly vulnerable to oxidative injury because of the overabundance of polyunsaturated fatty acids, which are susceptible to peroxidation by ROS.31,32 Protein modification is another route ROS uses to stimulate death. Specifically, ROS can degrade the antiapoptotic protein Bcl-2 by activating the ubiquitin protease system to cause beclin-1 activation and autophagy cell death.7,27,28 Oxygen free radical formation and lipid peroxidation enhance adverse mechanisms of neuronal injury, such as spinal cord hypoperfusion, development of edema, axonal conduction failure, and breakdown of energy metabolism.33,34 The importance of free radicals and lipid peroxidation in SCIs is supported by the large number of experimental and clinical studies demonstrating potential neural protective efficacy of pharmacological agents with antioxidant properties.
The Antioxidant Molecular Mechanism of Curcumin
Curcumin,1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione, or diferuloylmethane, the yellow pigment of turmeric and curry (Curcuma longa Linn), has been used in a variety of pharmaceutical applications as an antioxidant agent because it is easier to obtain and has fewer side effects than mitoquinone, a mitochondrial targeted antioxidant.35,36 In addition, curcumin has a stronger antioxidant capacity than vitamin E, resveratrol, and other antioxidants. 37 Curcumin not only reacts with ROS directly, but also acts as an activator of the antioxidant signaling pathway that is more extensive and lasting. 38 The mechanism of curcumin as an antioxidant could lie in its reaction with free radicals to generate phenoxy or carbon center free radicals on the methylene CH2 group. 19 These radicals are resonance stabilized and can be interconverted through conjugation. Curcumin has 2 phenolic O-H groups and 1 methylene CH2 group that are capable of H-bond dissociation enthalpy, whereas Trolox (a vitamin E analogue, in the presence of macromolecule-bound antioxidants in aqueous radical medium) has only 1 phenolic O-H. 39 Compared to Trolox, the presence of 2 identical OH groups as well as the methylene CH2 group in curcumin can easily undergo successive oxidations. 40 The free radical scavenging activity of curcumin arises by the resonance stabilization of its radicals from the 2 phenolic OH groups (mainly) or the CH2 group of the b-diketone moiety. 41 Thus, curcumin is not only a phenolic antioxidant that mostly donates H atoms from the phenolic groups but is a β-diketone radical chain-breaking substance that can give the H atom from methylene CH2. 37 Curcumin can treat free radicals by “electron transfer” and/or “H-atom donation.” Three different functional dissociation groups and a β-diketone site are important factors in curcumin’s antioxidant capacity, which has a better reduction capacity than Trolox.23,42,43 In cell membranes, curcumin intercepts lipid radicals and becomes phenoxy. Because phenoxy is more polar than curcumin, it moves to the membrane surface where it can be repaired by any water-soluble antioxidant, such as ascorbic acid. Therefore, curcumin, as a lipid radical scavenger, can protect cell membranes from oxidative damage.
The Therapeutic Effect of Curcumin in Injured Nerve Cells and Tissues
Curcumin has received much attention because of its antioxidant, anti-inflammatory, and angiogenesis effects.36,43 The therapeutic effects of curcumin have been evaluated in several diseases. In particular, studies have shown curcumin to inhibit the overactivation of microglia by regulating TLR4 expression in microglia, thereby reducing the inflammatory injury of neurons. 44 After curcumin pretreatment of transient global cerebral ischemic rats, 70% of cells in the hippocampal CA1 area had clear boundary, along with a stable karyotype, indicating that curcumin could protect nerve cells. 45 In the experimental model of autistic mice, curcumin promoted the proliferation of neural precursor cells, together with the differentiation and maturation of new neurons that enhanced the recovery of hippocampal neurons and alleviated autistic behavior of mice.46,47
Most investigative studies of curcumin relate to its antioxidant potentials. The biological classification of curcumin as both pro-oxidant and antioxidant is well supported by studies showing curcumin as a free radical scavenger, a reducing agent, and a DNA damage inhibitor. In vitro studies have shown curcumin to inhibit nitric oxide and ROS production in macrophages,4,48 whereas it also inhibited lipid peroxidation as well as cyclooxygenase in fibroblastic cells of rats 49 (Table 1).1,2,19,20,22,34,50-59
Therapeutic Effect of Curcumin in Injured Nerve Cells and Tissues.

Abbreviations: BDNF, brain-derived neurotrophic factor; CBP, CREB-binding protein; HO-1, heme oxygenase 1; MAPK, mitogen-activated protein kinase; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor κB; Nrf2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; CCI, chronic constrictive injury; PSR, phosphatidylserine receptor; STAT, signal transducer and activator of transcription; BCL, B cell lymphoma.
/ refers to No signaling pathway.
Curcumin Protects Against SCI by Scavenging Free Radicals Through the Nrf2/HO-1 Signaling Pathway
Owing to its anti-inflammatory,
60
antioxidative, and neuroprotective activities, curcumin has undergone investigative studies in SCI. For instance, the study by Zhang et al
25
evidenced curcumin’s potency in downregulating the expressions of N-methyl-
Curcumin can modulate autophagy and effectively ameliorate inflammation via activation of the Nrf2/HO-1 pathway and suppression of the NF-κB pathway in neurocytes. Also, curcumin can act as a scavenger of free radicals by stimulating the activity of HO-1 gene via the activation of the Nrf2/antioxidant response element (ARE) pathway during SCI repair. 62 Curcumin protects the white matter after SCI through inhibition of the NF-κB signaling pathway and activation of Nrf2 signaling by regulating ARE-driven antioxidant genes, such as HO-1.21,63 It is worth noting that HO-1 activity can lead to inhibition of NF-κB–mediated transcription through the action of bilirubin by decreasing free intracellular iron ions.64,65 Singh et al 66 reported the lack of TNF-α dependent phosphorylation and degradation of IκBα as well as the inhibition of translocation of the p65 subunit to the nucleus in curcumin-treated cells. Curcumin inhibits the NF-κB activation pathway at a step before phosphorylation but after the convergence of various stimuli. 67 Similarly, curcumin upregulated Nrf2 expression when NF-κB was inhibited by the inhibitor BAY 11-7082, whereas Nrf2 expression was significantly downregulated when the inhibitor was absent, 68 suggesting a possible interaction between NF-κB and Nrf2. Most particularly, curcumin could inhibit NF-κB and enhance Nrf2 expression, which could be paramount in the recovery of nerve-damaged tissue in the wake of SCI (Figure 2).
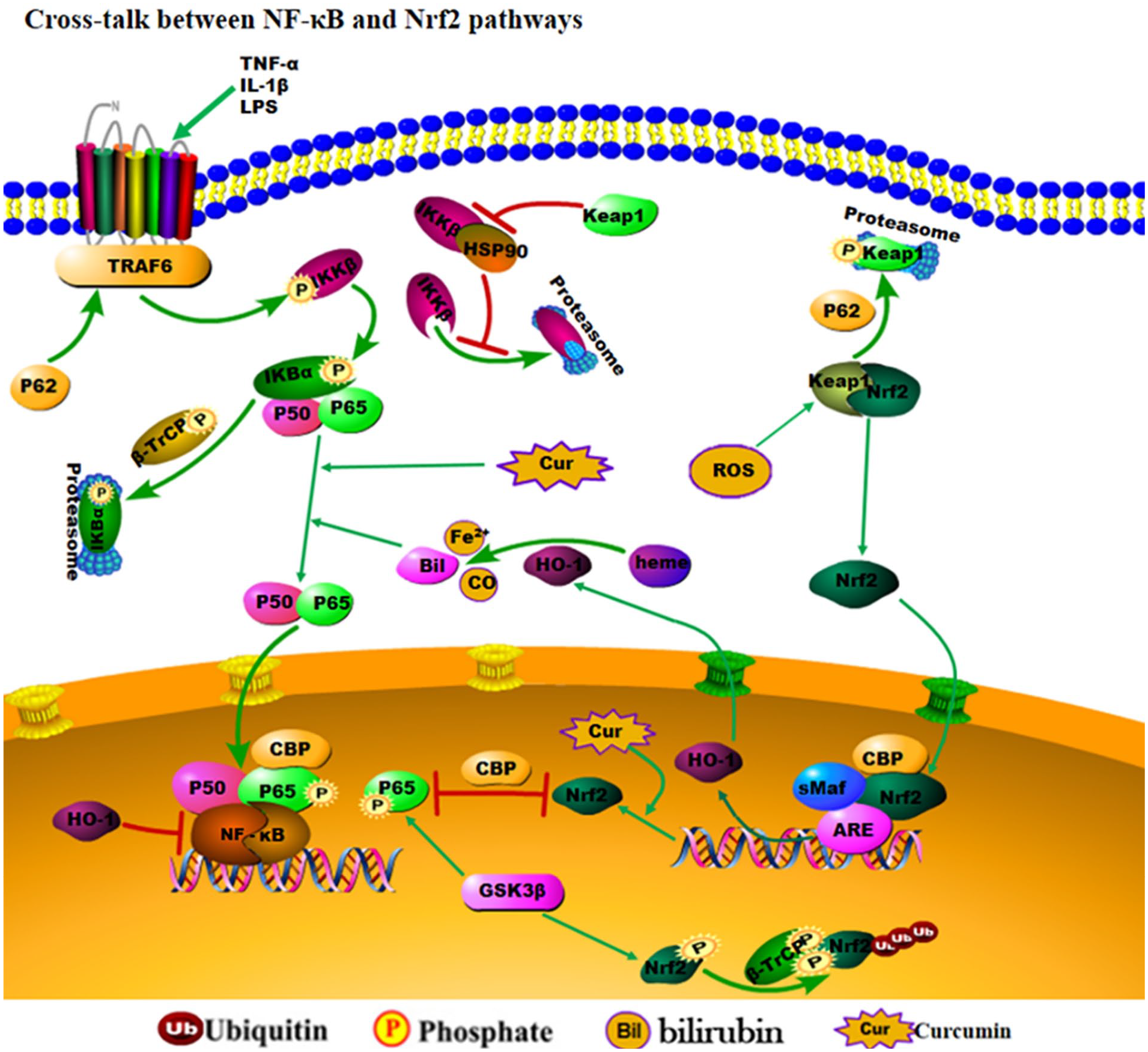
The cross talk between NF-κB and Nrf2 response pathways. At the transcriptional level, Nrf2 and NF-κB compete with transcription coactivator CREB binding protein. Keap1 depletes Nrf2 from the cytoplasm. However, the F-box protein, β-TrCP, controls nuclear Nrf2 levels. In the nucleus, β-TrCP promotes Nrf2 degradation by recognizing and binding to the Nrf2 phosphorylated by the GSK3β kinase. Interestingly, p65 is also a substrate for GSK3β phosphorylation. Moreover, β-TrCP is the regulation of IκBα degradation in response to cytokines. IKKβ is targeted for degradation through autophagy in the absence of HSP90. Keap1 can prevent HSP90 binding to IKKβ, which then triggers its autophagic degradation.
Conclusion
Free radicals and inflammation can lead to severe damage after SCI, and curcumin as a natural antioxidant possesses therapeutic properties for SCI. Curcumin can react with ROS to generate more polar phenoxy radicals, which are then transferred to the membrane surface for further hydrolysis. Furthermore, curcumin can activate Nrf2 to enhance the Nrf2/HO-1 signaling pathway and promote its antioxidant effect on free radicals and downregulate the NF-κB activation by inhibiting P65 to exert its anti-inflammatory function. Curcumin has a good outcome in alleviating inflammation and oxidative damage associated with SCI in mammals. In future clinical applications, a nanoparticle formulation necessitating the transport of curcumin to damaged areas of SCI could be used. However, for curcumin to be widely used in SCI treatments in humans, its biological characteristics and specific mechanism in nervous system injury and repair would warrant further extensive studies.
Footnotes
Authors’ Note
All authors agreed to publish this article. XL designed the study. WJ and XL prepared the first draft of the manuscript. BOAB, WJ, and XL revised the manuscript. All authors approved the final article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Zhejiang Province (No. LY19H170001) and the General research project of Zhejiang Provincial Department of Education (No. Y202044556).