
Abstract
Background
Agrin is a proteoglycan that aggregates nicotinic acetylcholine receptors (AChRs) on neuromuscular junctions and takes part in synaptogenesis in the development of the central nervous system. However, its effects on neural repair and synaptogenesis after stroke are still unclear.
Objective
This study aimed to investigate the effects of agrin on neural repair and synaptogenesis after stroke and the effects of exercise on this process in vivo and in vitro.
Methods
Exercise with gradually increased intensity was initiated at 1 day after middle cerebral artery occlusion (MCAO) for a maximum of 14 days. Neurological deficit scores and foot fault tests were used to assess the behavioral recovery. Western blotting, immunofluorescence, and electron microscopic images were used to detect the expression of agrin, synaptogenesis-related proteins, and synaptic density in vivo. In vitro, the ischemic neuron model was established via oxygen-glucose deprivation (OGD). The lentivirus overexpressed agrin and CREB inhibitor were used to investigate the mechanism by which agrin promoted synaptogenesis.
Results
Exercise promoted behavioral recovery and this beneficial role was linked to the upregulated expression of agrin and increased synaptic density. Overexpressed agrin promoted synaptogenesis in OGD neuron, CREB inhibitor downregulated the expression of agrin and hampered synaptogenesis in cultured neurons.
Conclusions
These results indicated that exercise poststroke improved the recovery of behavioral function after stroke. Synaptogenesis was an important and beneficial factor, and agrin played a critical role in this process and could be a potential therapeutic target for the treatment of stroke and other nervous system diseases.
Introduction
Cerebral ischemia (stroke) is the primary cause of death and long-term disability around the world, and most survivors of cerebral ischemia experience a combination of motor, cognitive, learning, memory, and language deficits.1,2 Unfortunately, to date, we still lack effective clinical approaches in the treatment of stroke. The recombinant tissue plasminogen activator (tPA), a thrombolysis agent currently used in clinical medicine, is limited by rigorous indications and a narrow therapeutic window. 3 Thus, it is urgent to establish an effective treatment strategy for improving neural repair and functional recovery after cerebral ischemia.
Growing evidence indicates that physical exercise is an effective therapeutic strategy for the prevention and treatment of stroke. Clinical investigations show that physical activity reduces the risk of cerebral ischemia. 4 Exercise rehabilitation after cerebral ischemia can promote the recovery of motor and sensory function and prevent muscular atrophy induced by denervation.5-7 Animal studies show that exercise is an effective therapeutic strategy in neuronal injury and diseases such as stroke, depression, 8 Parkinson’s disease, 9 memory deficit, and Alzheimer’s disease. 10 The results from our published studies suggest that exercise protects against ischemic brain injury and improves behavioral performance. 11 The underlying mechanisms include the reconstruction of the neurovascular unit 12 (neurogenesis and angiogenesis), promotion of mitochondrial biogenesis, 13 inhibition of acute inflammatory response 14 and apoptosis, 15 and protection of the blood-brain barrier. 16 Although exercise rehabilitation is a widely used therapeutic strategy and is supported by some clinical guidelines for stroke management,17,18 some recent reports have indicated that early exercise and very early mobilization (VEM) in patients with acute stroke was not associated with beneficial effects, and even reduced the odds of favorable outcome.19,20 These inconsistent results imply that currently there are no valid criteria in the formulation of an exercise program in clinical applications. Thus it is necessary to further explore the molecular mechanisms of exercise in the neural repair after stroke.
An impaired neural network is the major cause of dysfunction and disability in survivors from cerebral ischemia.21,22 Thus, the reconstruction of the neural network is the ultimate purpose of the treatment for cerebral ischemia, and the speed and degree of neural network reconstruction after cerebral ischemia is closely correlated with the recovery of neural function. 23 Synaptogenesis in the peri-infarct region is critical for the repair and reconstruction of the neural network. Agrin is a heparan sulfate proteoglycan that was originally identified in neuromuscular junctions.24,25 Agrin, which is secreted by motoneurons, is important to the formation and stabilization of neuromuscular synapses. 26 A recent study showed that the disruption of agrin expression led to microphthalmia and optic nerve hypoplasia in zebrafish embryos. 27 Also, agrin has been detected and is concentrated at synapses in the development of the central nervous system. 28 The dysfunction of agrin by knockdown technology inhibited synaptogenesis in cultured hippocampal neurons and slices.29,30 An in vivo study found that agrin-deficient mice die at birth because of dysplastic synapses, and perinatal death could be prevented by the expression of exogenous agrin in motor neurons. 31 Similarly, the results from Burk et al 32 indicated that agrin was necessary for the formation of mature synapses in the adult olfactory bulb. More recently, a study by Zhang et al 33 found that agrin was indispensable for neurogenesis and maturation in the hippocampus.
Together, these results indicate that agrin is an organizer and stabilizer in synapse formation during the development of the central nervous system. Similar to the development of the nervous system, synaptogenesis plays an important role in the process of repair of an injured neural network. However, the roles of agrin in synaptogenesis after cerebral ischemia are not well understood. In the present study, we investigated the effects of exercise on synaptogenesis and the roles of agrin during this process in vivo and in vitro. Our findings indicate that agrin involvement in synaptogenesis induced by exercise and could be a potential therapeutic target for the treatment of stroke and other nervous system diseases.
Methods
Animal and Group
All animal experiments were approved by the animal experimental committee of Yunnan University of Chinese Medicine at Kunming, China. Male Sprague-Dawley rats (260-280 g, Hunan SLAC Laboratory Animal Co Ltd) were housed under a 12:12 hour light/dark cycle at 21 ± 1 °C and were given free access to food and water. The rats were randomly divided into 3 groups: exercise with ischemia group (EI group), nonexercise with ischemia group (NI group), and sham group (without exercise and ischemia). Each group included 33 rats (12 used for Western blotting detection (day 7 and day 14 after middle cerebral artery occlusion (MCAO), n = 6 for each group), 12 used for immunofluorescence detection (day 7 and day 14 after MCAO, n = 6 for each group), 6 used for detection of infarct volume (day 7 after MCAO, n = 6 for each group) and 3 used for electron microscope detection (day 14 after MCAO, n = 3 for each group).
Middle Cerebral Artery Occlusion Model
In this study, transient MCAO was used to produce the experimental stroke model as previously described. 12 Briefly, all rats were anesthetized by 10% chloral hydrate (0.36 mL/kg intraperitoneally [i.p.]) and fixed in the supine position. The cervical skin was then incised and the left common carotid artery, the external carotid artery, and internal carotid artery were isolated successively. A small incision was made on the external carotid artery, a 4-0 nylon monofilament coated with a silicone tip (Shadong Biotech) was inserted into the origin of the middle cerebral artery from the incision on the external carotid artery. The ischemia was maintained for 60 minutes and then reperfusion was established by gently withdrawing the nylon monofilament. Rats in the sham group received all operation steps except for the occlusion of the middle cerebral artery. Physiologic variables (pH values, mean arterial blood pressure, paCO2, paO2, and rectal temperature) were monitored before, during, and after occlusion through the left femoral artery. A thermostat-controlled heating blanket was used to maintain the rectal temperature at 37.0 ± 0.5 °C during the operation.
Treadmill Training Protocol
The treadmill training protocol was performed according to our published article. 5 Briefly, all rats were habituated to the treadmill (Jide Experimental Instrument Co Ltd) at 6 to 9 m/min for 3 consecutive days (10 minutes per day) before the transient focal cerebral ischemia operation. Twenty-four hours after MCAO, the rats in the EI group received 7 or 14 consecutive days of exercise with gradually increased intensity according to a previously described protocol. 5 The intensity and duration of exercise was set as follows: on the first day, 5 m/min for the first 10 minutes, 9 m/min for the next 10 minutes, and 12 m/min for the last 10 minutes; on the second day, 5 m/min for the first 5 minutes, 9 m/min for the next 5 minutes, and 12 m/min for the last 20 minutes; on the 3rd to 14th day, 12 m/min for 30 minutes. The slope of the treadmill was set at 0° and the rats in the NI and sham groups were placed on treadmills with 0 m/min for 30 minutes. The time points for tests in the animal experiment are depicted in Figure 1. The rats in the NI and sham groups were placed on the electric treadmill for the same duration, but the velocity of the electric treadmill was set as 0 m/min.
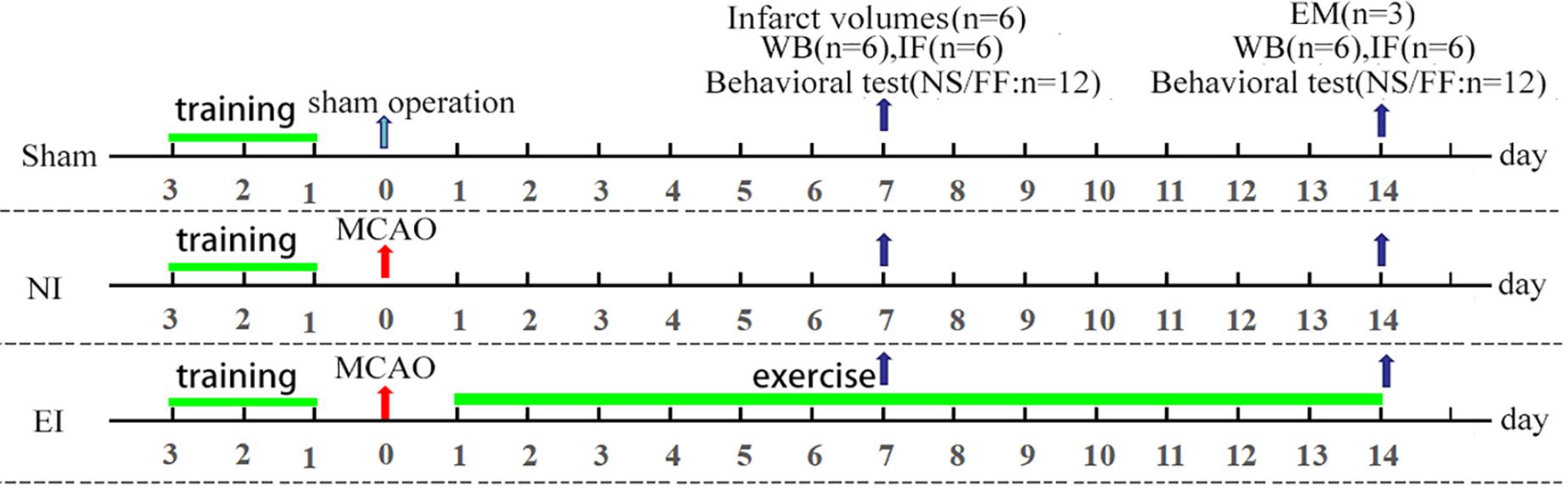
Schematic illustration of the experimental design in vivo. Sham, rats with sham operation; NI, nonexercise with ischemia; EI, exercise with ischemia; MCAO, middle cerebral artery occlusion; WB, Western blot; IF, immunofluorescence; NS, neurological score; FF, foot fault test; EM, electron microscopes; n, the number of rats in this test.
Behavioral Test
Neurological deficit scoring was used to assess the recovery of neurological defects induced by an experimental stroke at day 7 and day 14 after MCAO according to the previously described protocol. 34 The neurological deficit score was determined by an observer blinded to experiment design according to the following criteria: 0, no observable neurological symptoms; 1, flexion in the right forelimb; 2, reduced grip in the right forelimb; 3, flexion in the right forelimb, rotating toward the right side when held by the tail; 4, circling or walking to the right when walking; 5, failure to walk without help; 6, unable or difficult to ambulate spontaneously and 7, dead.
A foot fault test was used to assess the recovery of coordinated locomotor function of the affected forelimb (right forelimb) at day 7 and day 14 after MCAO, according to the previously described protocol.5,14 The placement of the right forelimb position on the rung was scored with a 7-point criteria: 0, total miss; 1, deep slip; 2, slight slip; 3, replacement; 4, correction; 5, partial placement; 6, correct placement. The higher score represented a better performance. The average scores of 3 trials were used for statistical analysis for each test day.
Detection of Brain Infarct Volume
Seven days after MCAO, 6 rats from each group were used to detect brain infarct volume. Briefly, rats were anesthetized with 10% chloral hydrate, the brains were quickly removed and cut into consecutive coronal sections with 2 mm thickness. These sections were then stained with 2% TTC (2,3,5-triphenyl tetrazolium chloride) solution for 30 minutes at 37 °C, followed by fixation in 4% paraformaldehyde buffer. The sections were photographed and the pale area was defined as the infarct zone, which was traced and calculated using NIH Image software. The percentage of infarct volume was determined according to the formula: infarct volume = (area of contralateral hemisphere − area of normal region in the ipsilateral hemisphere)/area of contralateral hemisphere×100%.
Protein Isolation and Western Blotting
Rats were sacrificed by deep anesthesia with 10% chloral hydrate (400 mg/kg, i.p.) at 7 days and 14 days after MCAO (see Figure 1). The cortical tissues in the ischemic penumbra (around the ischemic core, see Figure 3E) were isolated on ice. These tissues were homogenized in RIPA lysis buffer (Beyotime Biotechnology) for 40 minutes and centrifuged with 13 000 × g for 15 minutes at 4 °C. The total protein was harvested and quantified by bicinchoninic acid assay (BCA; Beyotime Biotechnology). Total proteins were separated on 10% sodium dodecyl sulfate–polyacrylamide gels and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore). The membranes were blocked in 5% w/v bovine serum albumin (Roche) for 1 hour at room temperature and then these blocked membranes were incubated in primary antibody against agrin (Millipore; 1:1000), synapsinⅠ (Cell Signaling Technology.; 1:1000), PSD95 (Abcam; 1:1500), BDNF (Abcam; 1:1000), CREB (Abcam; 1:500) and p-CREB (phospho S133, Abcam; 1:500), and beta actin (Sigma; 1:2000) for 24 hours at 4 °C. The next day, these membranes were washed 3 times in TBST (Tris-buffered saline containing 0.1% Tween-20) and incubated in horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Jackson Immuno-Research Laboratories, 1:2000) for 1 hour at room temperature. After three washes, the protein bands were detected with the Pierce ECL kit (Thermo Scientific) and semiquantified by fluorescence densitometry with the BIO-RAD system (Bio-Rad). The protein signals were normalized against the signals of beta actin.
Tissue Section Preparation and Immunofluorescence staining
Rats were deeply anesthetized with 10% chloral hydrate (400 mg/kg, i.p.) at 7 days and 14 days after MCAO (see Figure 1) and transcardially perfused with physiological saline and 4% paraformaldehyde. The brains were removed and dehydrated in 20% sucrose solution overnight (Invitrogen). Then the brains were cut into 30-µm-thick sections on a freezing microtome (SLEE). Thereafter, sections were permeabilized with 0.2% Triton X-100 for 15 minutes and were blocked with 10% normal goat serum for 1 hour at room temperature. Then sections were incubated with primary antibody against agrin (Millipore; 5 µg/mL), syn I (Abcam; 1:200), and PSD95 (Abcam; 1:500) for 1 hour at room temperature and then at 4 °C overnight. The next day, the sections were washed by phosphate-buffered saline (PBS) and incubated with DyLightTM 488- or 594-conjugated goat anti-rabbit secondary antibody (Jackson Immuno-Research Laboratories) for 1 hour at room temperature. After 3 washes, the sections were counterstained with DAPI (4′,6-diamidino-2-phenylindole) (Thermo Scientific) for 5 minutes followed by mounting with fluoromount-G (Southern Biotech, USA). The positive signals in the ischemic penumbra (around the ischemic core) were detected by a fluorescent microscope (Nikon Instruments Co, Ltd).
Transmission Electron Microscopy
Rats were deeply anesthetized and sacrificed with 10% chloral hydrate (400 mg/kg, i.p.) at 14 days after MCAO (see Figure 1). The brains were removed and the cortical tissue in the ischemic penumbra was isolated and cut into 1 mm blocks, and then fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 12 hours. Twelve hours later, these blocks were washed 3 times in 0.1 M PBS and fixed with 1% osmium tetroxide for 1 hour. Then the blocks were washed three times with pure water and dehydrated with ethanol. They were then embedded in epoxy resin and stained with uranyl acetate and lead citrate. Thereafter, ultrathin sections were produced with a Reichert ultramicrotome and pictured by a CM120 electron microscope at 80 kV.
Neuron Culture and Oxygen-Glucose Deprivation
Primary neurons were prepared from cortical tissues of 16- to 18-day embryos of Sprague-Dawley rats according to a published article
35
with some modifications. The pregnant female Sprague-Dawley rats were deeply anesthetized with isoflurane (Ruiwode) and sacrificed quickly. The embryos were obtained and put into precooling Dulbecco’s modified Eagle’s media (DMEM, Gibco) containing 10% fetal bovine serum (Lonza). The brains were quickly removed and the blood vessels and meninges were removed by tweezers under a stereo surgical microscope. The cortical tissues were removed from the brains of the pups and dissected into small blocks. These tissues were transferred to 15-mL conical tubes containing 0.5% trypsin (Gibco) and were incubated for 15 minutes in a 37 °C water bath. The supernatant was extracted and tissues were washed gently with precooling DMEM media containing 10% fetal bovine serum (Lonza). Cortical cells were dissociated by a sterile Pasteur glass pipette repeatedly and gently. The media containing dissociated cells were then static for 1 minute and the supernatant was used for cell counting. Without centrifugation, the cortical cells were seeded in poly-
The oxygen-glucose deprivation (OGD) was performed at 14 days of culture. During OGD, the primary neuron culture medium was replaced by basal culture media without glucose, and neurons were incubated in a hypoxia chamber supplied with 5% CO2 and balanced nitrogen for 2 hours, the oxygen level was less than 0.3% monitored by ProOx sensor (Biospherix). Two hours later, the neurons were returned to the normal culture condition. Twenty-four hours after OGD, cell death was detected by 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay (Beyotime Biotechnology).
Virus Transfection
The lentivirus-overexpressed agrin was produced by Cyagen Biosciences Inc. There were 3 lentiviruses named 1202, 1204, and 1205 and 1 control virus. After 3 days of culture, the lentivirus-overexpressed agrin was added in a culture medium. After 7 hours the culture medium added lentivirus was replaced by the primary neuron culture medium. Seventy-two hours later, the cell death and transfection efficiency were detected and the optimal multiplicity of infection (MOI) was chosen for the next experiment.
Treatment of CREB Inhibition
After 14 days of culture, the medium of the primary neuron was replaced by a medium containing 5- and 10-µm SGC-CBP30 (CREB inhibition, Selleck) respectively. Twenty-four hours later these cells were collected for detection.
Statistical Analysis
SPSS software (IBM Copr, version 17.0) was used for statistical analysis. All data were presented as mean ± standard error of the mean (SEM). Statistical differences were assessed by a 1-way analysis of variance (ANOVA) or Student’s t test. One-way ANOVA followed by post hoc Fisher’s PLSD (protected least significant difference) tests were used to determine statistically significant differences between EI, NI, and sham groups. Unpaired Student’s t test was used to compare behavioral data between the EI and NI groups. P value <.05 was defined as statistically significant.
Results
Physiological Variables and Mortality
There were no significant differences between groups for all the physiological variables, including pH, paO2, paCO2, and blood pressure before, during, or after MCAO among sham, NI, and EI groups (see Supplemental Materials). In the present study, we used an MCAO model with 1-hour ischemia, thus the mortality was low (4 of 70 rats in MCAO) and all of the deaths occurred within 12 hours after MCAO operation (before the randomization of NI and EI).
Exercise Increased the Expression of Agrin and Promoted Behavioral Recovery After Experimental Stroke
We detected the expression of agrin in the penumbra of the cortex, and the ischemic insult reduced the expression of agrin in the ischemic penumbra. Exercise increased the expression of agrin significantly (Figure 2A and C, 1.27 ± 0.06 in the EI group vs 0.73 ± 0.07 in the NI group, P = .0072 at day 7 of the training; 0.97 ± 0.07 in the EI group vs 0.87 ± 0.04 in the NI group, P = .0276 at day 14 of the training). Similarly, the results of immunofluorescence indicated that exercise increased the fluorescent spot compared with the rats in the NI group in the penumbra of the cortex at day 7 and 14 after MCAO (Figure 2B and D). Interestingly, the expression of agrin in the EI group was higher than the sham group at 7 days after MCAO (P = .0365). The same as the results of agrin, behavioral assessment confirmed that exercise promoted behavioral recovery of the neurological deficit (Figure 2E, 2.08 ± 0.34 in the EI group vs 3.14 ± 0.43 in the NI group, P = .0300 at day 7 of the training; 1.33 ± 0.26 in the EI group vs 2.25 ± 0.39 in the NI group, P = .0387 at day 14 of the training) and coordinated locomotor function compared with the rats in NI group (Figure 2F, 4.65 ± 0.26 in the EI group vs 3.59 ± 0.19 in the NI group, P = .0352 at day 7 of the training; 5.13 ± 0.14 in the EI group vs 4.05 ± 0.18 in the NI group, P = .0366 at day 14 of the training). Additionally, exercise significantly reduced the infarct volume in the cortex and striatum (Figure 2G and H, 31.62% ± 2.16% in the EI group vs 46.78% ± 1.37% in the NI group, P = .0232). These findings suggested that exercise after experimental stroke might significantly increase the expression of agrin and promote behavioral recovery after experimental stroke.
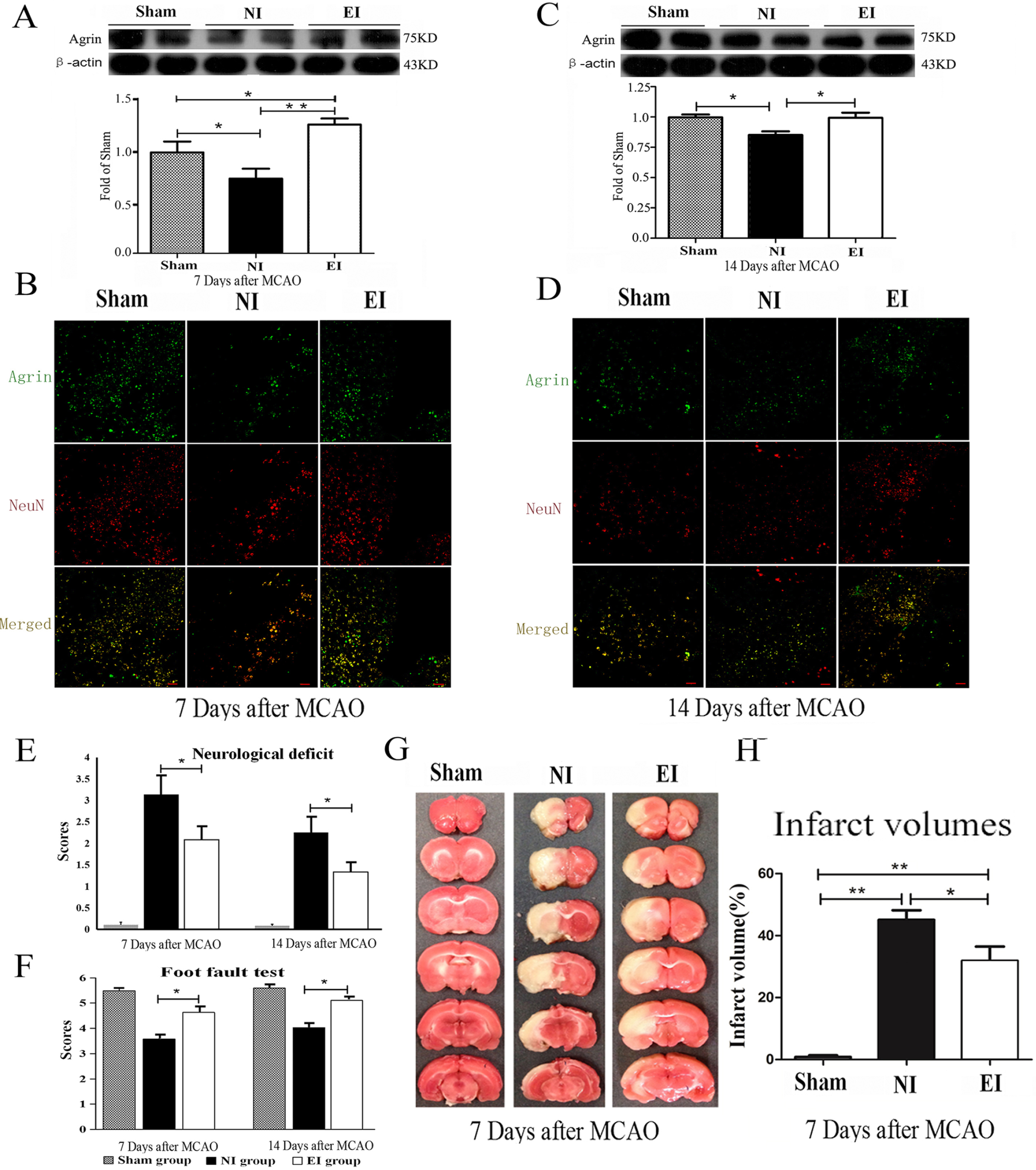
Exercise improved behavioral recovery and upregulated the expression of agrin. Protein bands and semiquantitative results of agrin in ischemic penumbra detected by Western blotting at 7 and 14 days after middle cerebral artery occlusion (MCAO), respectively (A and C). Representative images of agrin-stained sections at 7 and 14 days after MCAO, respectively (B and D). Scale bar, 100 μm. Quantitative results of neurological deficit score and foot fault test (E and F). Representative images of TTC (2,3,5-triphenyl tetrazolium chloride)-stained section (G) and quantitation of the infarct volume (H). n = 6 for each group in A, B, C, D, G, and H; n = 12 for each group in E and F; *P < .05, **P < .01.
Exercise Increased Synaptic Densities in Ischemic Penumbra
Agrin induces dendritic filopodia and synapse formation in the brain. Its up-regulated expression might be related to synaptogenesis. To verify this hypothesis, we detected synaptogenesis induced by exercise with Western blotting, immunofluorescence, and transmission electron microscopy. The results showed that exercise increased the expression of pre-synaptic proteins synapsin I (Figure 3A, 1.18 ± 0.09 in the EI group vs 0.55 ± 0.21 in the NI group, P = .0281 at day 7 of the training; 0.96 ± 0.01 in the EI group vs 0.83 ± 0.05 in the NI group, P = .0425 at day 14 of the training) and postsynaptic density protein 95 (PSD95) (Figure 3C, 1.08 ± 0.06 in the EI group vs 0.74 ± 0.09 in the NI group, P = .0063 at day 7 of the training; 1.04 ± 0.07 in the EI group vs 0.68 ± 0.16 in the NI group, P = .0383 at day 14 of the training) at days 7 and 14 after MCAO, respectively. Similarly, the results of immunofluorescence confirmed the results of Western blotting (Figure 3B and D). The ultrastructure detected by transmission electron microscopy indicated that ischemic insult severely damaged synapses and the neural network. Exercise increased the number of synapses(16.15 ± 0.85 in the EI group vs 10.77 ± 0.74 in the NI group, P = .0385) and the potential functional synapses (the synapses containing synaptic vesicles) (8.16 ± 0.76 in the EI group vs 3.14 ± 0.42 in the NI group, P = .0197) (Figure 3G-J). These results indicate that exercise after experimental stroke might improve synaptogenesis in the ischemic penumbra.
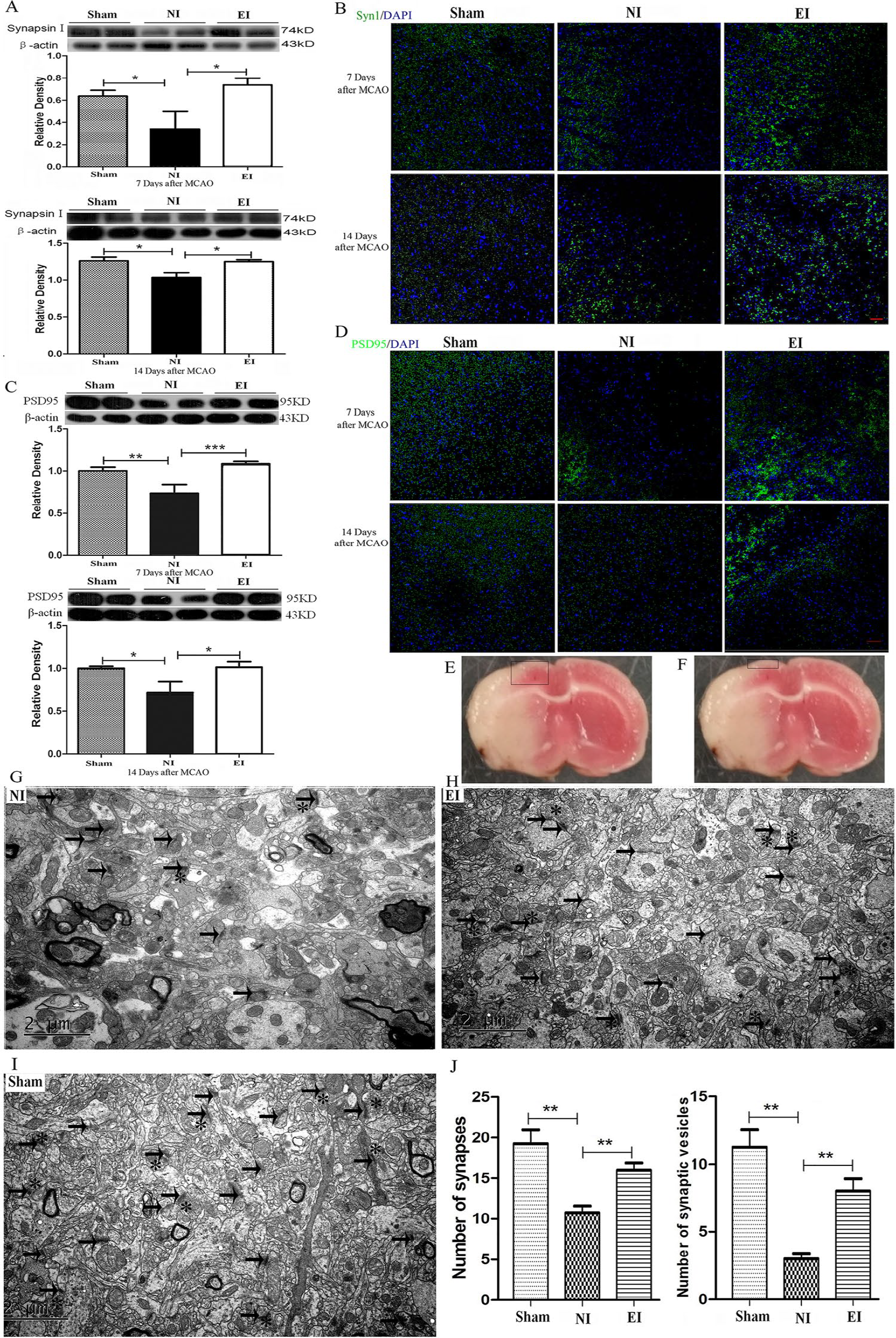
Exercise increased synaptic density. Protein bands and semiquantitative results of presynaptic marker synapsin I and postsynaptic marker PSD95 (postsynaptic density protein 95) in ischemic penumbra detected by Western blotting at 7 and 14 days after middle cerebral artery occlusion (MCAO), respectively (A and C). Representative images of synapsin I and PSD95 stained sections at 7 and 14 days after MCAO, respectively (B and D). Scale bar, 100 μm. The diagram shows where the tissue was sampled in Western blotting and the where photomicrographs were taken in immunofluorescence staining (E) and where the tissue was sampled in transmission electron microscopy (F). Representative transmission electron microscopy (G-J), arrow: synapse in ultrastructure, *: synaptic vesicle, scale bar, 2 μm. Quantitative analysis of synapse and synaptic vesicle (H). n = 6 for each group; *P < .05, **P < .01.
Exercise Increased the Expression of Synaptogenesis-Related Proteins
Neurotrophic factors and growth-related proteins are essential to synaptogenesis. We furthermore determined the expression of these proteins, including brain-derived neurotrophic factor (BDNF), growth-associated protein 43 (GAP43), and cAMP-response element binding protein (CREB, a key transcription factor in growth and development). The results indicated that exercise markedly upregulated the expression of BDNF (0.98 ± 0.05 in the EI group vs 0.78 ± 0.07 in the NI group, P = .0357 at day 7 of the training; 0.99 ± 0.02 in the EI group vs 0.82 ± 0.03 in the NI group, P = .0281 at day 14 of the training) and GAP43 (1.36 ± 0.03 in the EI group vs 1.16 ± 0.05 in the NI group, P = .0086 at day 7 of the training; 0.92 ± 0.06 in the EI group vs 0.66 ± 0.09 in the NI group, P = .0263 at day 14 of the training) at days 7 and 14 after MCAO, respectively (Figure 4A and B). The expression of both total CREB (0.97 ± 0.02 in the EI group vs 0.80 ± 0.07 in the NI group, P = .0433 at day 7 of the training; 0.99 ± 0.05 in the EI group vs 0.84 ± 0.03 in the NI group, P = .0359 at day 14 of the training) and phosphorylated CREB (1.06 ± 0.01 in the EI group vs 0.82 ± 0.04 in the NI group, P = .0351 at day 7 of the training; 0.94 ± 0.05 in the EI group vs 0.76 ± 0.07 in the NI group, P = .0420 at day 14 of the training) were higher in the EI group compared with the NI group (Figure 4C and D). These findings imply that exercise increased the expression of neurotrophic factors and growth-related proteins, and promoted the synaptogenesis in ischemic penumbra.
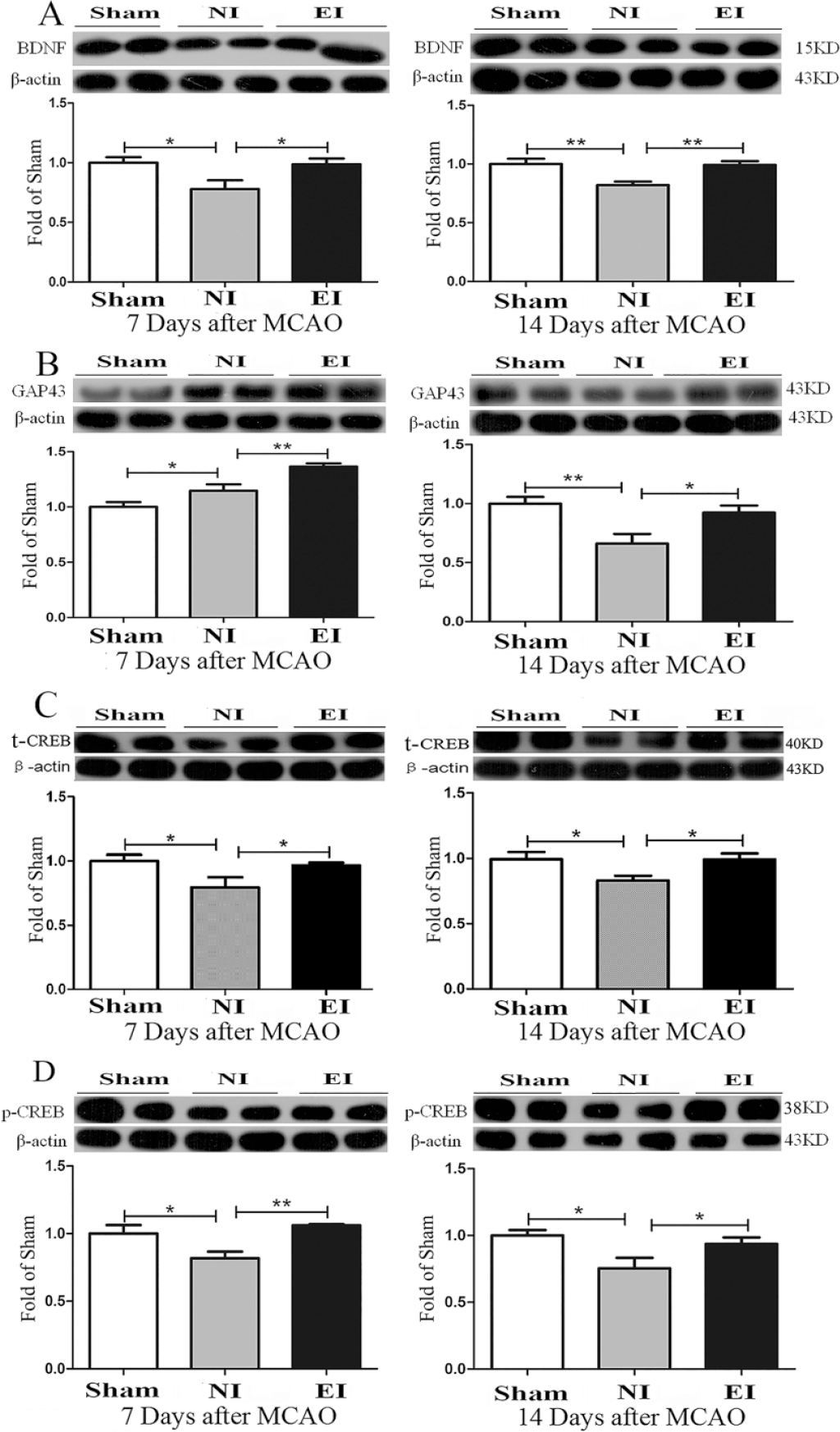
Exercise increased the expression of synaptogenesis-related proteins. Protein bands and semiquantitative results of BDNF (A), GAP43 (B), CREB (C), and p-CREB (D) in ischemic penumbra detected by Western blotting at 7 and 14 days after middle cerebral artery occlusion (MCAO), respectively. n = 6 for each group; *P < .05, ** P < .01.
Overexpressed Agrin Promoted Synaptogenesis in OGD Neurons
In order to explore the effects of agrin on the synaptogenesis in ischemic neurons, we overexpressed agrin in OGD neurons via lentivirus with the agrin gene. Our findings showed that overexpressed agrin in injured neurons upregulated the expression of synapsin I (a marker for presynaptic element, 2.08-fold of sham, P = .0159) and PSD95 (a marker for postsynaptic element, 1.63-fold of sham, P = .0237) (Figure 5C-E). Accordingly, the expression of BDNF was increased in neurons with overexpressed agrin (1.70-fold of sham, P = .0256, Figure 5F). These results indicated that overexpressed agrin promoted synaptogenesis in OGD neurons.
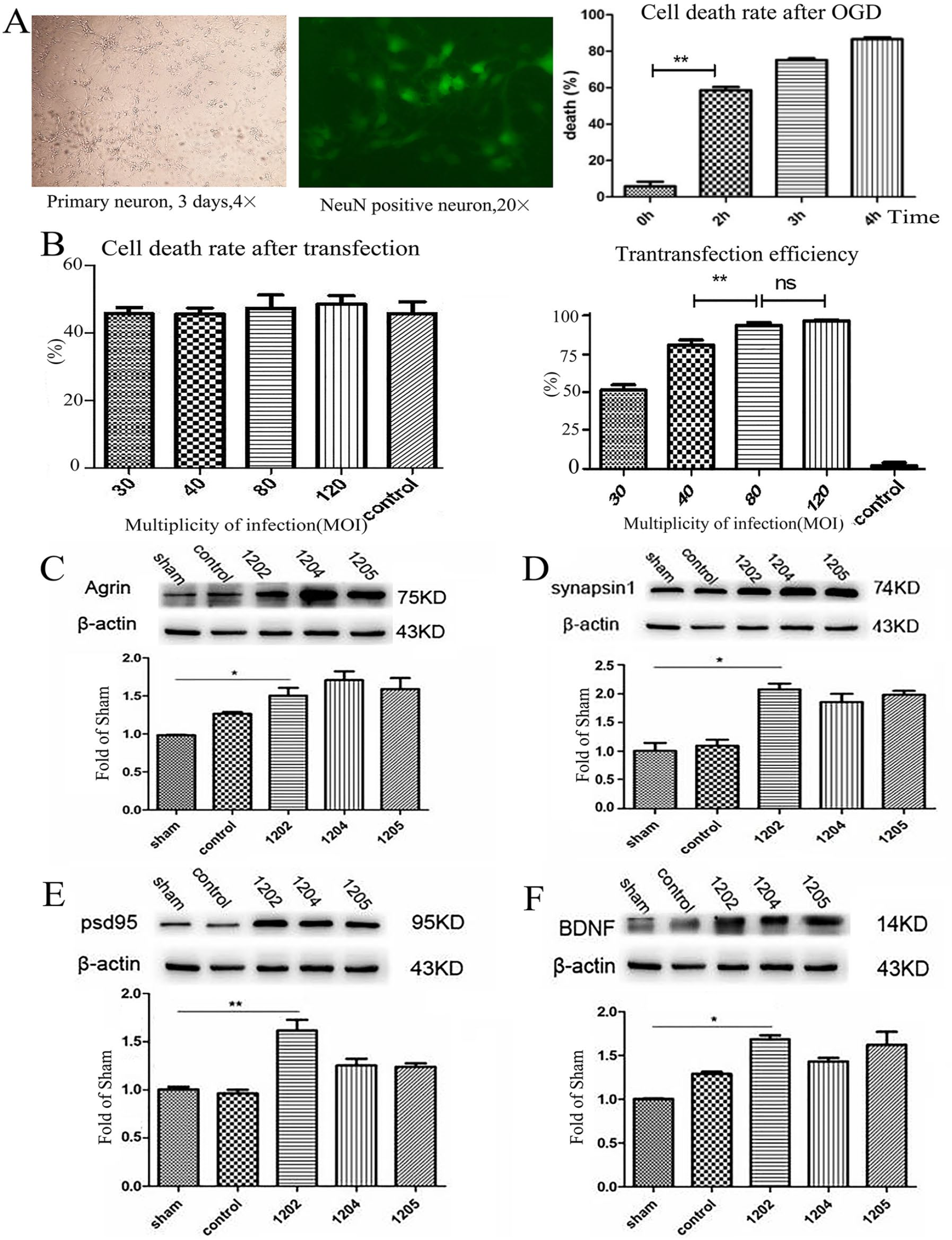
Overexpressed agrin promoted synaptogenesis in oxygen-glucose deprivation (OGD) neuron. Primary neuron isolated from cortex of 16- to 18-day embryos of Sprague-Dawley rat (A left) and cell death rate after OGD (A right). Lentivirus-mediated transfection of agrin (B and C). Overexpressed agrin upregulated the expression of presynaptic marker synapsin I (D), postsynaptic marker PSD95 (E), and BDNF(F) in OGD neuron, respectively. n = 5 for each group; *P < .05, **P < .01.
Inhibition of CREB Disrupted Synaptogenesis in OGD Neuron
The phosphorylation of CREB is an important upstream regulator for CREB/BDNF signaling pathway and plays an important role in neuroprotection and neural repair. Our results showed that the CREB inhibitor SGC-CBP30 (Selleck) markedly downregulated the expression of BDNF (0.83 ± 0.03 fold of sham in the SGC-CBP30 with 5 µm, P = .0320), agrin (0.72 ± 0.05 fold of sham in the SGC-CBP30 with 5 µm, P = .0416), and synaptic biomarker synapsin I (0.27 ± 0.10 fold of sham in the SGC-CBP30 with 5 µm, P = .0062) and PSD95 (0.17 ± 0.04 fold of sham in the SGC-CBP30 with 5 µm, P = .0038) (Figure 6C-F). These results indicated that the CREB pathway could be the upstream regulator of agrin and the target of the synaptogenesis following the brain injury.
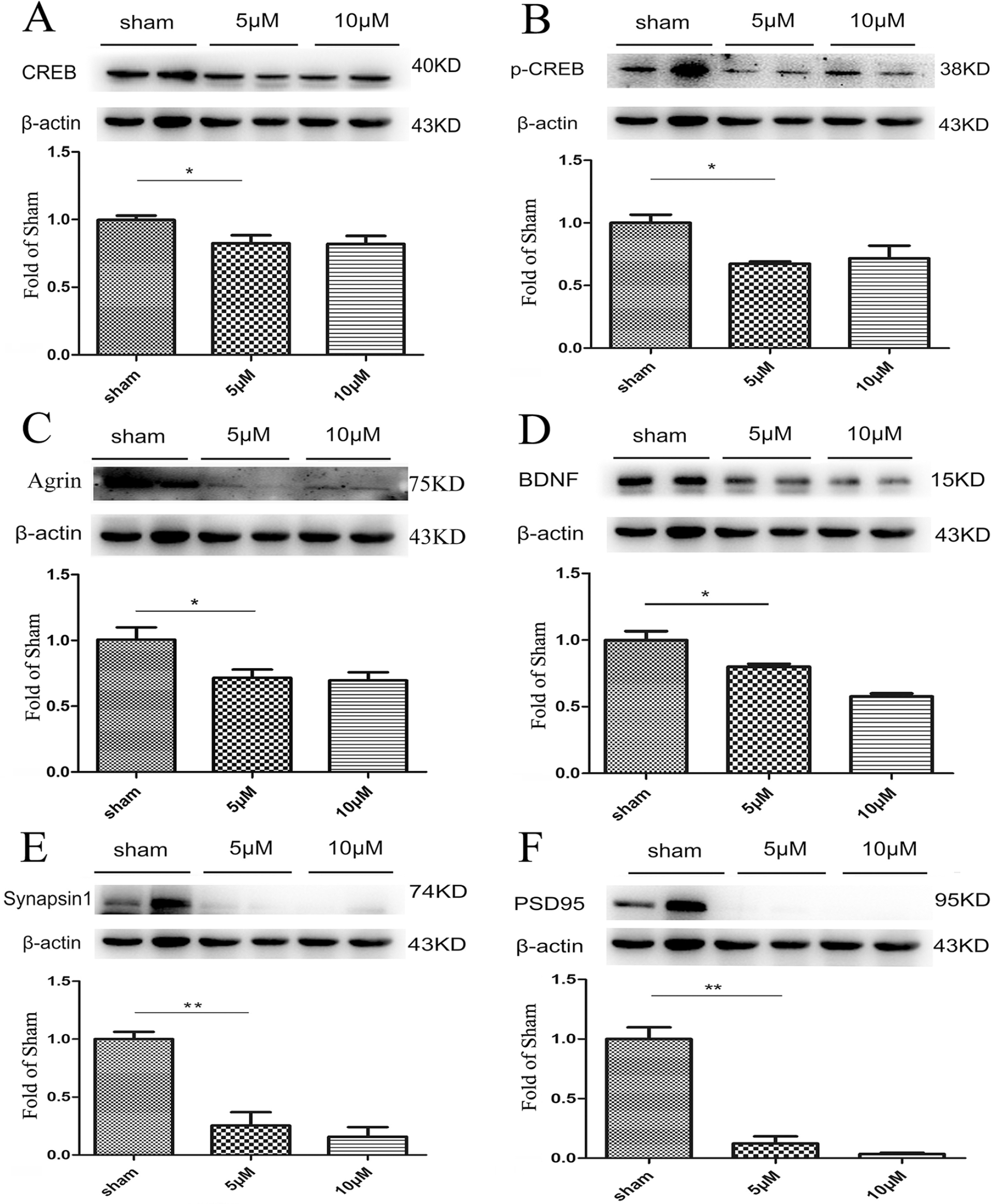
Inhibition the CREB disrupted synaptogenesis in oxygen-glucose deprivation (OGD) neuron. The CREB inhibition SGC-CBP30 downregulated the expression of total CREB (A) and phosphorylated CREB (B) in 5 μM and 10 μM. The inhibition of CREB downregulated the expression of agrin (C), BDNF (D), presynaptic marker synapsin I (E) and postsynaptic marker PSD95 (F), and in OGD neuron, respectively. n = 5 for each group; *P < .05, **P < .01.
Discussion
The destruction of the neural network is the direct and disastrous consequence in brain injury, such as stroke, trauma, cerebral palsy, neurodegenerative disorders, and so on. The reconstruction of the neural network is the ultimate purpose for treatment of brain injury. The synapse is the core of the neural network, thus the pathways and proteins involved in synaptogenesis will be potential treatment targets for brain injury. In the present study, by in vivo and in vitro experiments, we found that agrin played an important role in synaptogenesis and that exercise could increase the expression of agrin and promote synaptogenesis in a rat model of experimental stroke.
The brain is a critical organ with high plasticity potential. The neural network is broken following stroke and the brain initiates the reparative process and attempts to reconstruct the neural networks. 24 However, spontaneous repair is insufficient and limited by many factors. The reconstruction of the neural network is the ultimate purpose of treatment of stroke. 25 The plasticity in brain injury is different from regeneration in an injured spinal cord: it occurs in a complex environment that includes neural stem cells, injured neurons (surviving bodies and dead axons or synapses), denervated intact neurons, and glia. The repair processes following brain injury includes endogenous neurogenesis, axon regeneration, myelination, synaptogenesis, and reconstruction of the neural network.36-38 A thorough clarification of the cellular and molecular mechanisms of plasticity in brain injury and the neural network reconstruction is a necessary endeavor in the search for new therapeutic targets for brain injury.
Although it has been confirmed that agrin has an organizing role in synapse formation and stabilization in neuromuscular junctions, the effect of agrin on the repair of the central nervous system injury is unclear and seldom studied.39-42 In the present study, we observed that recovery of function improved by exercise was accompanied by an upregulated expression of agrin in a rat model of stroke, and we confirmed that overexpressed agrin promoted synaptogenesis in cultured OGD neurons. Moreover, agrin contributed to the reconstruction of the neurovascular unit and improved the microenvironment of nerve regenerative niche by stabilizing the adherens junction proteins in brain endothelial cells. 43 A recent report confirmed that agrin took part in adult hippocampal neurogenesis stimulated by an enriched environment through the Agrin-Lrp4-Ror2 pathway. 33 Together, these findings imply that agrin is involved in neural network reconstruction and could be a potential therapeutic target in treatment for cerebral ischemia and other brain injuries. However, the underlying mechanisms of neural plasticity and synaptogenesis induced by agrin are unclear. The upstream regulator of agrin includes some noncoding RNAs such as miR-144. 44 The verified downstream factors of agrin, myogenic muscle-specific receptor tyrosine kinase (MuSK), are lower in expression in the brain and Lrp4 could be the specific receptor of agrin in the brain.33,45 In addition, agrin promotes angiogenesis in cancer progression, which may imply that agrin can improve the neural plastic niche and contribute to sypaptogenesis. 46
Physical exercise is an efficient and natural way to enhance neurogenesis and synaptogenesis in both the normal and injured brain. A recent study from the Kai Chen group indicated that chronic treadmill exercise increased dendritic spines, promoted oligodendrogenesis and axonal myelination, and improved motor learning function through activating the mechanistic target of the rapamycin (mTOR) pathway in mouse motor cortex. 47 Studies from 2 groups (Aimone et al 48 and Tashiro et al, 49 respectively) indicated that exercise promoted the maturation of synapses in the adult hippocampus, but the increased maturation was dependent on the density of dendritic spines or branching. 50 Some clinical researchers confirmed that exercise mitigated white matter atrophy 51 and improved white matter microstructure in the corpus callosum. 52 Our preliminary results showed that exercise improved the micro-environment of neurogenesis and synaptogenesis through inhibiting neuroinflammatory responses, increasing angiogenesis following cerebral ischemia, and reducing neural apoptosis and infarct size.12,14 Here we observed that exercise after cerebral ischemia increased the expression of a synaptogenesis-related protein and the number of synapses via Western blotting, immunofluorescence, and electron microscopes. Importantly, for the first time, we found that the increased synaptogenesis induced by exercise was accompanied by an upregulated expression of agrin.
Thus, in addition to the beneficial effect of neuronal plasticity induced by exercise, the neuroprotection induced by exercise also contributes to recovery after ischemia. Functional recovery from stroke is correlated with multiple factors, which include neuroprotection (suppression of apoptosis, neuroinflammation, etc) and neuronal plasticity (angiogenesis, neurogenesis, etc). Although they may come into play at different times, the synergistic effects of neuroprotection and neuronal plasticity contribute to the reconstruction of neural networks and functional recovery.
Exercise benefits have been confirmed repeatedly and applied in clinical practice for many years, however, it is not easy to choose the type of exercise: forced or voluntary exercise training. Forced exercise, such as treadmill training, is a proven, controllable, and effective rehabilitation protocol in the treatment of stroke.11,18 Also, forced exercise is beneficial to the rehabilitation of cardiopulmonary function after stroke. However, unsuitable forced training may cause systemic stress and muscle strain.53-55 On the other hand, voluntary exercise such as wheel running in animal studies induces less corticosterone stress response than forced training 56 and may lead to a better emotional experience and more sustainable improvement.57,58 Voluntary exercise is uncontrollable in clinic application and leads to less neuroprotective effects compared with forced treadmill training.59,60 Thus the application of exercise in a clinical setting should consider various factors including sex, age, lifestyle, blood-brain barrier disruption, risk factors, complications, individual differences, and so on. The underlying mechanisms of neuroprotection and neuronal plasticity induced by exercise need to be clarified and incorporated into the basis of the exercise protocol design.
Although we confirmed the beneficial effects of agrin in synaptogenesis in this study, the underlying mechanisms need to be further confirmed in vivo. Additionally, we did not investigate different exercise parameters such as initiation time, period, intensity, and type of exercise in this study. The underlying mechanisms of agrin induced by exercise need to be further clarified in the future.
Conclusion
The present study demonstrates that exercise after stroke improved recovery of motor function. Synaptogenesis is an important and beneficial factor, and agrin plays a critical role in this process and could be a potential therapeutic target for the treatment of stroke and other nervous system diseases. Further work is required to explore the underlying molecular mechanisms.
Supplemental Material
Table_1._The_physiological_parameters_among_different_groups_before,_during,_and_after_ischemia – Supplemental material for Agrin Involvement in Synaptogenesis Induced by Exercise in a Rat Model of Experimental Stroke
Supplemental material, Table_1._The_physiological_parameters_among_different_groups_before,_during,_and_after_ischemia for Agrin Involvement in Synaptogenesis Induced by Exercise in a Rat Model of Experimental Stroke by Pengyue Zhang, Liqiang Yang, Guangxiang Li, Yaju Jin, Danli Wu, Qing Mei Wang and Peidong Huang in Neurorehabilitation and Neural Repair
Footnotes
Supplementary material for this article is available on the Neurorehabilitation & Neural Repair website along with the online version of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (81660384, 81960731, 81460351), Yunnan Applied Basic Research Projects (2017FB119), and Yunnan Province University Innovation Team Projects (“Acupuncture Prevents Mental Disease”).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.