
Abstract
Background and objective. Prior studies have suggested that after stroke there is a time-limited period of increased responsiveness to training as a result of heightened plasticity—a sensitive period thought to be induced by ischemia itself. Using a mouse model, we have previously shown that most training-associated recovery after a caudal forelimb area (CFA) stroke occurs in the first week and is attributable to reorganization in a medial premotor area (AGm). The existence of a stroke-induced sensitive period leads to the counterintuitive prediction that a second stroke should reopen this window and promote full recovery from the first stroke. To test this prediction, we induced a second stroke in the AGm of mice with incomplete recovery after a first stroke in CFA. Methods. Mice were trained to perform a skilled prehension (reach-to-grasp) task to an asymptotic level of performance, after which they underwent photocoagulation-induced stroke in CFA. After a 7-day poststroke delay, the mice were then retrained to asymptote. We then induced a second stroke in the AGm, and after only a 1-day delay, retrained the mice. Results. Recovery of prehension was incomplete when training was started after a 7-day poststroke delay and continued for 19 days. However, a second focal stroke in the AGm led to a dramatic response to 9 days of training, with full recovery to normal levels of performance. Conclusions. New ischemia can reopen a sensitive period of heightened responsiveness to training and mediate full recovery from a previous stroke.
Introduction
Most training-associated recovery at both impairment and functional levels occurs in the first month after stroke in rodent models1,2 and in the first 3 months after stroke in humans.3-5 We have previously shown that the medial agranular cortex (AGm), a medial premotor area, reorganizes after a focal stroke in the caudal forelimb area (CFA), rodent primary motor cortex, and mediates recovery if poststroke training is initiated after a 1-day poststroke delay (ie, 48 hours after stroke) but not if initiation of training is delayed for a week.6,7 We refer to this period of increased responsiveness to training as the poststroke sensitive period, which represents a unique, time-limited environment of heightened plasticity 8 —that is, the sum of molecular, physiological, and structural changes that lead to greater behavioral gains for the same amount of training inside as compared with outside the sensitive period.2,8
Experiments in rodents suggest a causal link between the unique short-lived plasticity milieu after stroke and the amount of recovery from hemiparesis in this same period. For example, manipulation of plasticity in the sensitive period either by increasing9,10 or decreasing brain-derived neurotrophic factor (BDNF) 11 augments or prevents recovery, respectively. Furthermore, Nudo et al 12 demonstrated that training monkeys on skilled digital retrieval of food pellets from small wells after an infarct in the hand area of the primary motor cortex prevented loss of hand representation in the peri-infarct cortex. In contrast, withholding motor training led to a decrease in digit representations by more than 50%.12,13 Thus motor training directs reorganization in remaining cortical areas, including premotor areas, presumably enabled by the unique poststroke plasticity milieu.
A counterintuitive implication of a stroke-induced sensitive period is that a second stroke should reopen a sensitive period and thereby trigger recovery from a prior stroke. That is, if ischemia heightens plasticity, motor deficits induced from a first stroke could be reversed by repeat ischemic damage if rehabilitative training is initiated at an appropriate time. To test this idea, we induced a second focal stroke in the medial premotor area of mice that had only partially recovered prehension (reach-to-grasp) performance after a first CFA stroke because motor training had been delayed. We then induced a second stroke and retrained the mice after a 1-day delay (ie, 48 hours after stroke). The prediction was that training within the sensitive period induced by the second stroke would lead to full recovery from the first stroke.
Material and Methods
Subjects
Adult male C57bl/6 mice 100 to 150 days old were singly housed in custom-made chambers and kept on a 12/12-hour light/dark cycle. Behavioral tasks were carried out in the same room and same chambers in which the mice were housed to reduce the stress of new surroundings. Two to 3 days prior to learning the prehension task, mice were placed on a scheduled administration of 2.5-g Bio-Serv dustless precision pellet mouse chow per day with water ad libitum. Mice were food restricted to 85% of their starting weight. We studied a total of 15 mice based on effect sizes in previous studies.1,6,14 Three mice were excluded from the analysis: 2 died prior to completion of the study; the other was excluded because of failure to induce a stroke in the AGm. Mice were randomized to, and investigators blinded to, training condition. All animal handling and use was performed according to the protocols set by the Johns Hopkins University Animal Care and Use Committee.
Skilled Prehension Task
Training was conducted as described previously. 6 Briefly, standard mouse cages were modified with a sealable, vertical 16 cm × 0.9 cm slit through which the mice would stick their paw, as well as with a standing steel stage measuring 1.5 cm × 11.5 cm × 8 cm directly in front of the slit. Once the mice were familiarized to the pellets and had lost 15% of their body weight, they were trained on the prehension task. For this, 45-mg Bio-Serv dustless precision pellets were placed on sticky tape on a movable steel bar and maintained at the same height as the standing steel cage. The pellets were positioned 0.5 cm away from the standing steel stage and aligned with the edge of the cage slit contralateral to the preferred paw. This configuration required the mouse to reach and grasp with its preferred paw for pellets one at a time. Prehension was scored as a success when the mouse reached its forelimb through the slit, grabbed the pellet, and ate it without knocking it from its resting space, dropping it, or in any other way losing control of it. Otherwise, the attempt was recorded as a miss. Our behavioral outcome measure was percentage of successful prehension attempts; if the mouse did not touch the pellet, it was not counted as an attempt. Paw preference was determined in a series of preliminary reaching blocks that were not scored. Once paw preference was determined and the mice were familiar with the task, the space between the opening of the cage and bar loaded with food pellets was increased to a maximum distance of 1 cm to increase the difficulty of the task. The mice then underwent 2 blocks of 30 reaching attempts per training day. The animals had 1 training day off per week (including the day after stroke induction). During training, mice were also fed at the end of each day with additional food pellets placed in their cage to maintain their weight at 85% of baseline. Training after stroke began either 48 hours or 8 days after stroke induction and followed the same protocol as described above. All investigators were blinded to training condition after stroke induction.
Stroke Induction
The location of motor areas was identified based on prior anatomical 15 and functional 16 mapping. These areas are geographically consistent within a given strain, and we have used them with prior success.6,7 Focal cortical infarction was induced by photothrombosis of the cortical microvessels with some modification to previously described protocols. 17 Each mouse was anesthetized with 4.5 mL/kg of a ketamine (21 mg/mL) plus xylazine (3.2 mg/mL) mixture and placed in a stereotaxic frame (Stoelting, Wood Dale, IL). Temperature was monitored and maintained at 36.5°C to 37.5°C with the help of a heating pad. At the dorsal aspect of the head, the skull was exposed by a median incision of the skin; the periostium was removed and the bregma point identified. The skull was thinned using a fine dremmel. A fiber optic bundle of a cold light source (Zeiss 1500 electronic, Jena, Germany) with a 20-gauge aperture was centered at 2 mm lateral and 0.5 mm anterior from bregma (CFA 16 ), 0 mm laterally and 0.5 mm anteriorly from bregma (AGm), or 2.5 mm laterally and 1 mm anterior of lambda (visual cortex) and placed against the skull. The brains were then illuminated through the intact skull for 15 minutes starting 5 minutes after the IP injection of 150 μl of a 10-mg/mL rose Bengal solution in sterile normal saline. The scalp was then sutured, and mice were allowed to awaken while still on the heating pad. Dual location and size (≈0.25 mm3) of stroke was pathologically confirmed in each mouse included in the analysis; there was no difference in size of the strokes (data not shown).
Statistics
The effect of the second stroke was analyzed with a repeated-measures ANOVA, with 3 relevant time points (just prior to stroke 1, just prior to stroke 2, and the last training day) as within-group factors and the second stroke location as the between-group factor. Post hoc comparisons made with Sidak’s correction.
Results
To test if a second stroke could reopen a sensitive period and allow full recovery from a first CFA stroke, we followed the experimental paradigm shown in Figure 1A. Specifically, using custom-built cages (schematized in Figures 1B to 1E), we trained wild-type adult male mice to perform a skilled prehension task to an asymptotic level of performance, photothrombotically induced a focal CFA infarction (t1; Figures 2A and 2B), and retrained them after a 7-day delay. Assessment on the prehension task on day 18 (8 days after stroke) revealed that there was little spontaneous recovery of performance, which is in agreement with our prior results, 7 and subsequent training over 19 days in total led to only mild performance gains, which never returned to prestroke levels (Figures 3A and 3B, days 18-36). A second focal stroke was then induced in the ipsilesional medial premotor area (AGm) at time point t2 (Figures 2A and 2B). Training was started after a 1-day poststroke delay (ie, 48 hours after stroke). After t2, prehension performance was initially worse, as would be expected from a second stroke in an ipsilesional premotor area 6 but then returned to the normal level seen before the first CFA stroke (Figure 2A; t3). As a control, another group of mice was given a second stroke in the ipsilesional occipital cortex. This group showed neither significant worsening nor subsequent improvement. There was no significant between-group difference in prestroke performance at t1 (2 tailed, paired t test), although this could be a result of lack of power. This is not a problem, however, because the control group started, if anything, at a higher level of performance than the premotor group (although, again, this difference did not reach significance via a 2-tailed, paired t test). Nevertheless, despite starting from this higher level of performance, the control group did not show any recovery after a second stroke in the occipital cortex, whereas a second stroke in the medial premotor area led to a dramatic reversal of the motor deficit caused by the first stroke (Figures 2B and 2C).
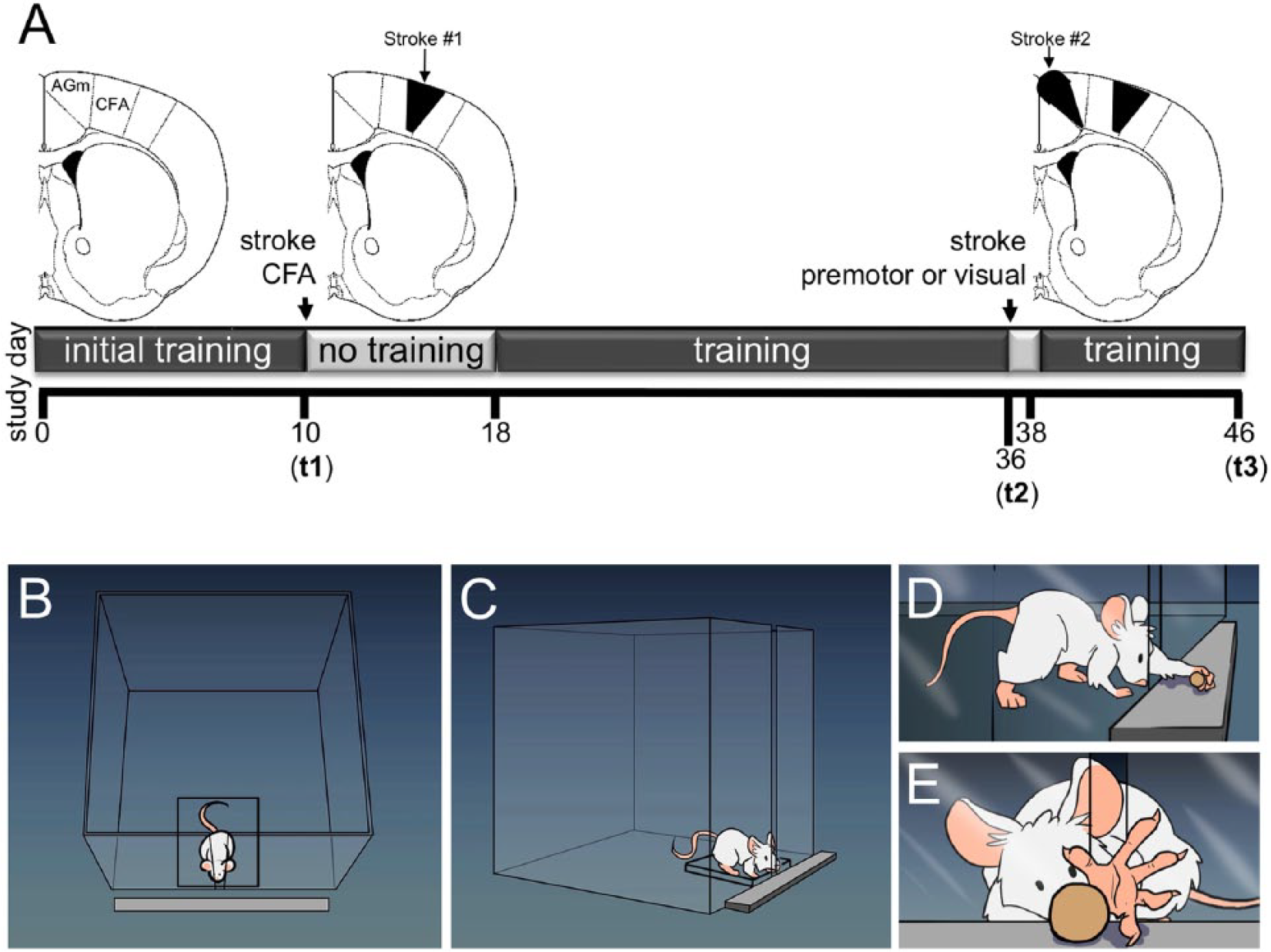
(A) Schematic of experimental timeline: initial CFA stroke at t1; second stroke, which was induced in either the medial premotor area (AGm) or the visual cortex (occipital lobe) at t2; the mice were sacrificed at t3. (B-E) Cartoon of prehension task and training apparatus.
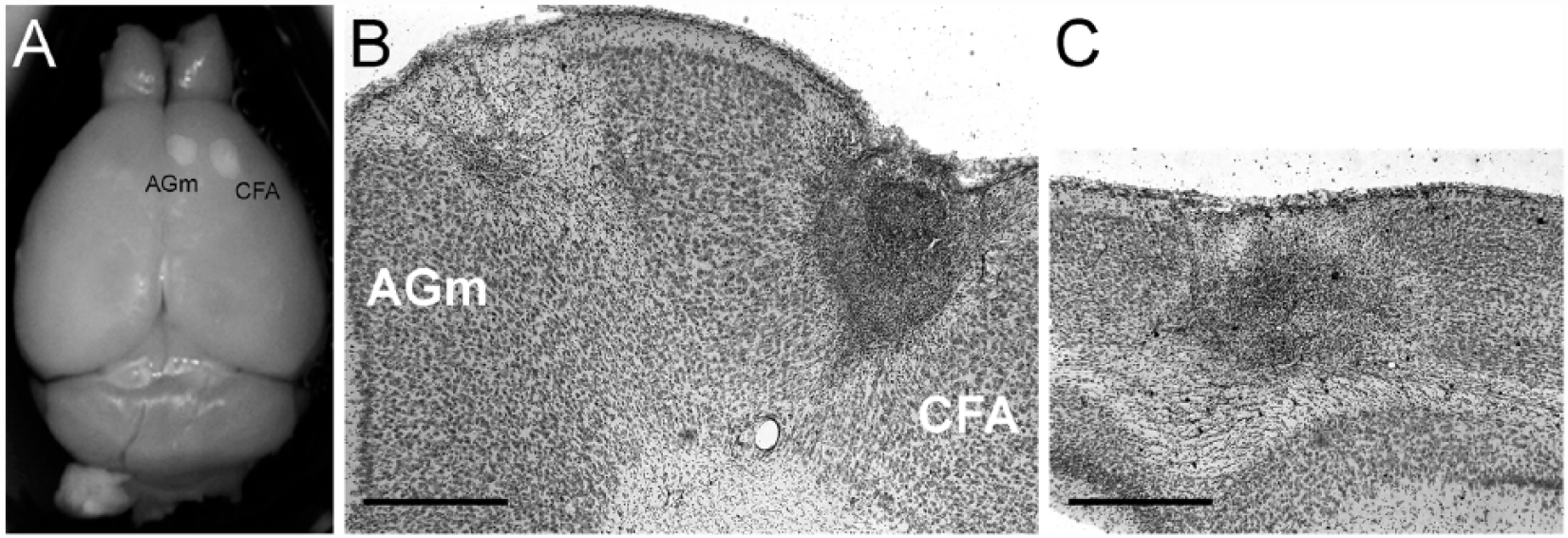
Representative images of CFA, AGm, and occipital strokes: (A) Paraformaldehyde-fixed brain with strokes in CFA and AGm. (B) Representative 50-μm Cresyl violet–stained coronal sections through the frontal lobe, showing strokes in both the CFA and AGm. (C) Representative 50-μm Cresyl violet–stained coronal sections through the occipital lobe showing a stroke in the visual cortex. Scale bars = 200 μm.
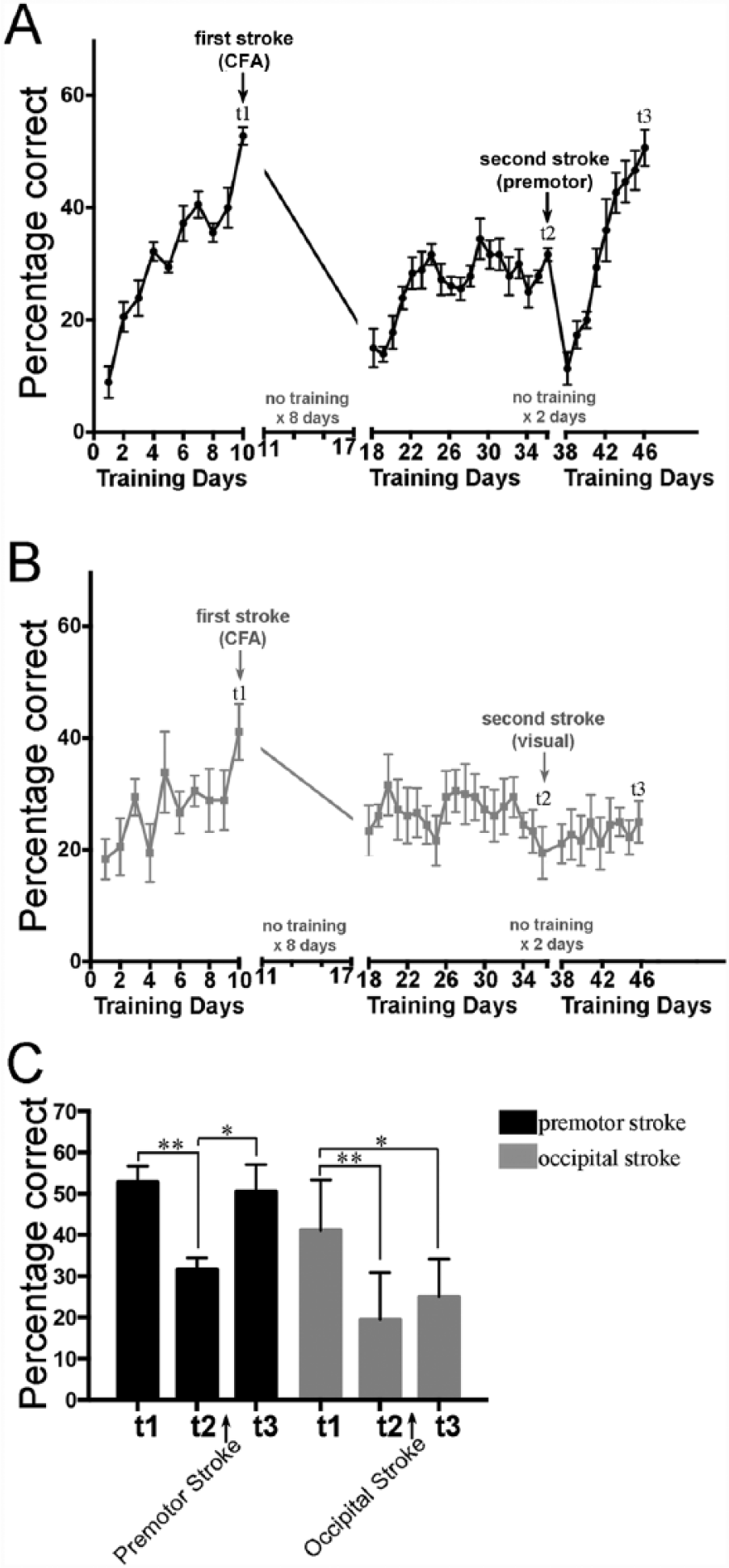
Mice were trained to perform the skilled prehension task to an asymptotic level of performance (t1), after which they underwent photocoagulation-induced stroke in the caudal forelimb area (CFA). After a 7-day poststroke delay, the mice were retrained for 19 days. A second photocoagulation-induced stroke was then induced in either the ipsilesional medial premotor cortex (A) or in the ipsilesional visual cortex (B) at time t2. The mice were retrained after only a 1-day delay (ie, 48 hours later) and killed humanely at t3. Each group comprised 6 mice. (C) Prehension performance at time points t1, t2, and t3. A repeated-measures ANOVA showed a significant interaction between group and time points t1, t2, and t3 (P = .015). Asterisks indicate significant post hoc differences compared using Sidak’s multiple comparisons test (*P < .001; **P < .0001).
Discussion
Here we show that a second stroke in the ipsilesional premotor cortex was sufficient to reopen a sensitive period and restore responsiveness to training after it had reached a plateau following a first stroke in the primary motor cortex, with subsequent full recovery. This is the first example of a double-stroke paradigm being used to reopen a poststroke sensitive period and induce motor recovery.
The Poststroke Sensitive Period
Recent work in rodent models has shown that there are unique molecular, structural, and physiological changes in the peri-infarct cortex during the first 4 weeks poststroke that likely serve as the substrate for increased plasticity in response to training.2,8 Many of these peri-infarct changes peak within the first 7 days8,18,19 and then begin to normalize. The lack of responsiveness to further training (eg, during days 18-36) that we saw here in the mouse is consistent with studies in the rat that failed to show a benefit of late tune-ups despite a response to training early after stroke. 20 That the impact of training falls off rapidly within 1 week poststroke is consistent with our prior results 7 and also consistent with results in rats showing only a modest response to training and enrichment given 2 weeks poststroke, compared with when given early. 1 Human data also suggest a short-lived plasticity window after stroke with most spontaneous recovery, which follows a predictable proportionality rule,3,21 occurring in the first 3 months.4,22
The results reported here show that mice had the capacity to recover back to normal even when they had hit a performance plateau and were no longer responding to training after the first CFA stroke (ie, days 22-36). Poststroke performance reached a plateau by approximately day 23—that is, after 4 days of training. There was no further improvement despite 14 extra days of training. After a second stroke in the AGm, there was a marked transient worsening in performance, but it then improved beyond the previous plateau within 4 days (day 42) and continued to improve for the next 5 days after this. Thus, whereas the mice showed a flat training response for 14 days before the second stroke, after it they showed an immediate and sustained steep training response for 9 days. In stark contrast, a second stroke in the visual cortex led to no training response over 9 days. These observations make it highly implausible that the mice, in the absence of the second motor stroke would have suddenly started to dramatically respond to training on days 38 and 39 despite a complete lack of responsiveness for the previous 2 weeks or that the mice with a second stroke in the visual cortex would have suddenly started to respond dramatically had they continued beyond 9 days. Instead, the only plausible explanation for our results is that the renewed responsiveness to training after an apparent performance plateau was reached is attributable to a second stroke in the AGm. We chose the AGm because in prior work, we showed that it reorganizes after CFA stroke. 6 It is possible that a stroke in the rostral forelimb area (RFA) or another premotor area may have produced similar results.
These results have important implications for understanding mechanisms of recovery early after stroke. In a primate model of stroke, the amount of reorganization in the ventral premotor cortex was proportional to the size of the ischemic lesion in the primary motor cortex, suggesting a dose-like response to factor release after ischemia. 23 In a rodent model, it has been shown that AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor modulation starting at day 5 after stroke further augments the increases in BDNF expression seen in the peri-infarct cortex and is associated with improved motor recovery. 24 Conversely, antagonizing tonic GABA (γ-aminobutyric acid) inhibition in the peri-infarct cortex early after stroke enhances motor recovery. 25 Thus, manipulation of the peri-infarct milieu during the sensitive period can augment and/or prolong the heightened plasticity that transiently occurs and also mitigate those factors that begin to normalize it.
There are obvious similarities between the poststroke sensitive period and developmental critical periods during which environmental stimuli are most likely to influence brain reorganization. 26 Perhaps the best examples of environment-induced plasticity come from studies of the visual system, in which there are limited time windows within which specific visual stimuli can alter gene expression, dendritic spine dynamics, neuronal tuning, and ultimately circuit connectivity.27,28 Of particular interest are those studies that show how visual cortical critical periods can be reinstated in the adult rodent.29-32 Similar to our results, these studies indicate that the adult brain has the capacity to undergo large-scale change in response to environmental stimuli but requires some additional stimulus (eg, fluoxetine) to overcome inhibitory mechanisms. 7
Double Lesions and Recovery
The double-lesion approach has been used in previous studies to identify those regions mediating recovery after a first stroke. In these cases, the second lesion is used to reinstate the original deficit, not reverse it as we did here. For example, in a previous study, we showed that the medial premotor area undergoes reorganization after training-induced recovery from CFA stroke. 6 Similarly, a second stroke placed in the RFA after rats had been rehabilitated from an initial CFA stroke reinstates the initial stroke-induced phenotype.33,34 Conversely, it has also been shown that CFA can mediate recovery after a focal RFA stroke. 34 Thus previous double-lesion experiments have been used to probe reorganization rather than induce it. In addition, these prior studies did not directly test the importance of rehabilitation timing after stroke. Specifically, there was either no rehabilitation after the second stroke, 33 or rehabilitation was initiated with the same delay used after the first stroke. 34
The double-lesion effect that we observed is also distinct from those paradoxical lesions that physiologically ameliorate symptoms from a primary neurological condition. For example, bradykinesia resulting from degeneration of the nigrostriatal pathway (caused by ischemia or otherwise) can be improved by lesions (including stroke) in the internal pallidal segment or in the subthalamic nucleus. 35 In this case, the mechanism of such improvement is fast, independent of rehabilitation, and is caused by a reduction of excessive and disordered activity in the inhibitory pallido-thalamic pathway.36-38 This physiological mechanism is quite distinct from a slower training-dependent effect in a period of heightened plasticity.
Finally, the double-lesion effect that we observed is distinct from ischemic preconditioning, in which initial sublethal cerebral ischemia results in the upregulation of certain genes, which conveys tolerance to later, otherwise lethal, ischemia. Thus, ischemic preconditioning lessens the impact of a stroke by decreasing infarct size.39-41 In contrast, our second stroke increased infarct volume and initially worsened the residual deficit from the first stroke. It was only after training that behavioral recovery was seen.
Implications for Recovery in Humans
Whereas previous work has shown that the sensitive period can be pharmacologically modulated once it is already in progress,7,42 we show that the poststroke sensitive period can be reset once it is over. Although this proves the existence of a postischemic sensitive period, inducing a second stroke is not a tenable therapeutic option for patients. Recent work, however, has shown that there may be other ways to reset critical periods in the healthy adult rodent using certain molecules, medications, and/or modification of inhibitory/excitatory balance.29-32 Such approaches may provide insight into how to reset the sensitive period in the absence of an additional ischemic insult.
Footnotes
Acknowledgements
The authors wish to acknowledge Boone Snavely for his illustrations.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr Zeiler is currently supported by 1K08NS085033-01, the Richard S. Ross Clinician Scientist Award as well as by a startup fund from the Johns Hopkins Department of Neurology. Dr Krakauer is currently supported by R01 NS052804–05, R01 120 86264, R01 HD073147, James S. McDonnell Foundation 220020220.