
Abstract
Hoffman OR, Koehler JL, Espina JEC, Patterson AM, Gohar ES, Coleman EM, Schoenike BA, Espinosa-Garcia C, Paredes F, Varvel NH, Dingledine RJ, Maguire JL, Roopra AS. Sci Transl Med. 2025;17(790):eadt0527. All current drug treatments for epilepsy, a neurological disorder affecting more than 50 million people, merely treat symptoms, and a third of patients with epilepsy do not respond to medication. There are no disease-modifying treatments that may be administered briefly to patients to enduringly eliminate spontaneous seizures and reverse cognitive deficits. Applying network approaches to whole tissue and single-nucleus transcriptomic data collected from mouse models of temporal lobe epilepsy and publicly available transcriptomic data from human temporal lobectomy samples, we confirmed a previously described pattern of rapid and transient induction of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway within days of epileptogenic insult. This was followed by a resurgent activation of the JAK/STAT pathway weeks to months later with the onset of spontaneous seizures. Targeting the first wave of JAK/STAT activation after epileptic insult did not prevent seizures. However, inhibition of the second wave with CP690550 (tofacitinib) over a 2-week period enduringly suppressed seizures, rescued deficits in spatial memory, and alleviated epilepsy-associated histopathological alterations. Seizure suppression lasted for at least 2 months after the final dose. These results indicate that reignition of inflammatory JAK/signal transducer and activator of transcription 3 signaling in chronic epilepsy opens a window for disease modification with the US Food and Drug Administration-approved, orally available drug CP690550.
Commentary
The holy grail in epilepsy research is finding a treatment that can reduce seizures, improve comorbidities, and has few side effects. Although rarely considered possible, it would be ideal to taper off medications after seizures stop and continue to have seizure freedom. In their work, the authors may have achieved those goals. 1
The authors started by trying to understand whether a histone methyltransferase called enhancer of zeste homolog 2 (Ezh2) can suppress epileptogenesis, following up on prior studies implicating the molecule in rodents and human temporal lobe epilepsy (TLE). 2 To advance their understanding they crossed a mouse with floxed Ezh2 with a mouse that had Cre-recombinase in cells with the synapsin 1 promoter, leading to a deletion of Ezh2 in mature neurons. They compared these mice to wild-type (WT) controls in a mouse model of TLE. This model uses the convulsant kainic acid to induce several hours of severe, continuous seizures, called status epilepticus (SE). After SE, the animals develop spontaneous recurrent seizures, usually following a delay of several weeks.
It is known that many genes are up- or down-regulated after SE in a variety of neuronal and nonneuronal cells. Theoretically, if one could identify the main drivers of these changes, it might be possible to prevent epileptogenesis, a strategy that already has some support. 3 Therefore, the authors set out to try to find the transcription factors or cofactors that might be responsible for most changes in gene expression after SE.
They started with hippocampal samples 1 day after SE and used RNA-seq. They identified a cluster of genes that were expressed much more in the mice that had SE and Ezh2 deleted compared to WT mice. Afterward, they used gene ontology analysis, which identified the innate immune response. They were particularly interested in what the analysis called “Interleukin-6 (IL6) Janus kinase (JAK) STAT3 (Signal transducer and activator of transcription 3) and tumor necrosis factor signaling” because it has been previously implicated in epilepsy.4-6 The authors had also shown before that STAT3 increases gene expression in models of epilepsy. 2
They then used a tool developed by the group termed Mining Algorithm for Genetic Controllers, which showed that STAT3 was the top candidate for control of gene expression in their cluster. Furthermore, STAT3 was a central node in the gene regulatory network (GRN). Other analyses of the GRN, such as betweenness centrality, implicated STAT3 as well. STAT3 is interesting because it is known to be phosphorylated by JAKs, mainly JAK1. Phosphorylated STAT3 (pSTAT3) dimerizes and then translocates to the nucleus to exert its effects on target genes (Figure 1A), often genes related to the immune system and inflammation, but also cell proliferation and other functions. 8 The JAK/STAT pathway can refer to other JAKs (JAK1, 2, 3, or Tyk2) and STATs (1, 2, 3, 4, 5a, 5b, or 6). 9
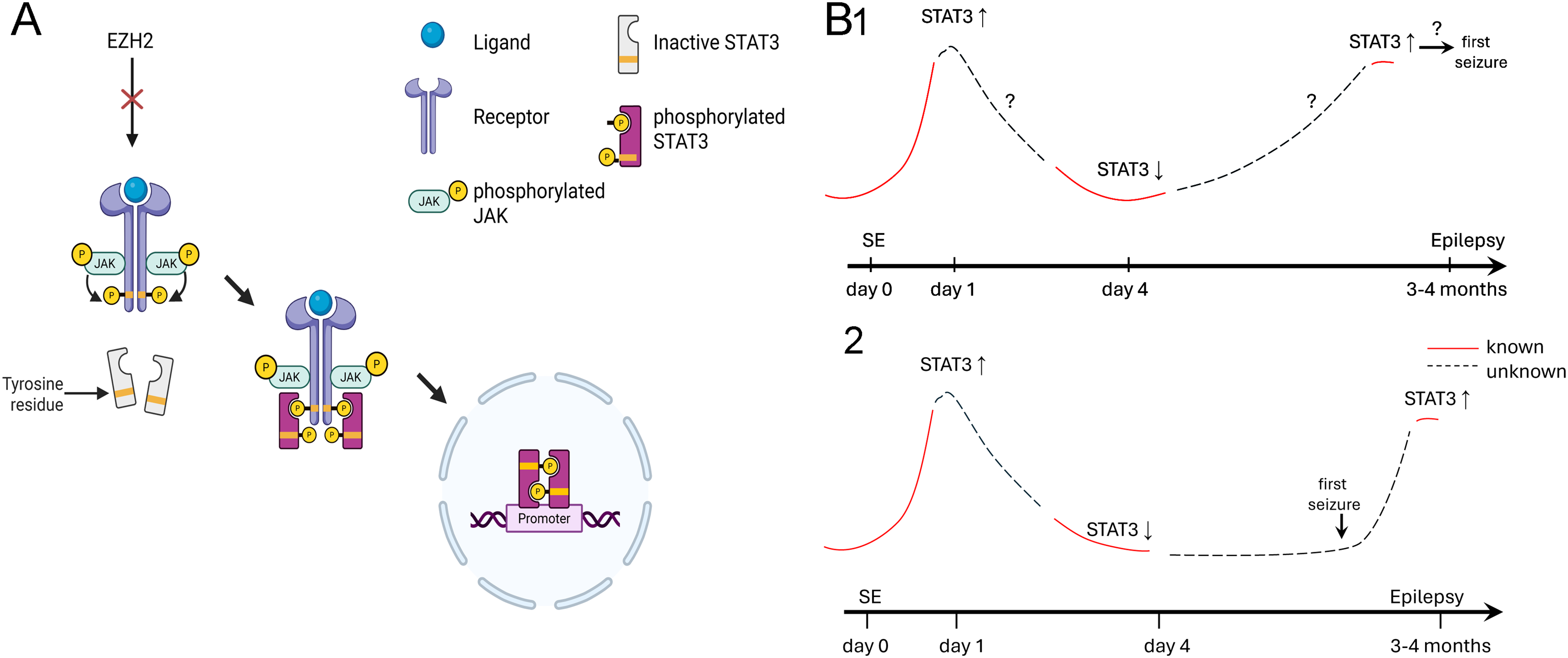
STAT3 signaling and its resurgence during epileptogenesis. (A) A diagram of the JAK/STAT pathway is shown with Ezh2 as a prominent regulator. After binding of ligands such as cytokines and growth factors, the receptor for the ligand is phosphorylated by JAK, making the receptor a docking site for STAT3. JAKs phosphorylate STATs, leading to their dimerization and translocation to the nucleus where they act on target genes. Created in BioRender. 7 . (B) A diagram of the mouse model of TLE used by the authors to study levels of STAT3. SE was induced and 1 day later STAT3 levels were high, followed by a decline by day 4. After 14 weeks they found STAT3 was elevated again, suggesting a resurgence. (1) One explanation of resurgence is that during epileptogenesis something occurs that triggers an elevation of STAT3 and that initiates the first seizure. Here STAT3 plays a pivotal role in the transition to spontaneous recurrent seizures. (2) Another explanation is that the first seizure during the phase of chronic epilepsy triggers an elevation of STAT3, which might recur every time there is a seizure. Here STAT3 is not necessarily critical but simply associated with the chronic phase of epilepsy.
The next question was whether STAT3 was increased after SE. They used western blots to show that STAT3 was elevated 1 day after SE, as were other members of the JAK/STAT pathway, which confirmed prior findings. Interestingly, when samples were analyzed 4 days instead of 1 day after SE, pSTAT3 was no longer elevated. The results are reminiscent of numerous studies of genes upregulated after SE that seem to return to normal within days, leading some to question whether studying these genes would be fruitful. Fortunately, the authors analyzed a later time, 14 weeks after SE. They found an upregulation of STAT3 as well as JAK1. They called this resurgence a “reignition,” which is a very important observation. The idea of a reignition is a conceptual advance because it suggests a new aspect of epileptogenesis that has not been widely appreciated. Instead, epileptogenesis in TLE has been suggested to progress linearly after the initial insult or possibly sigmoidally, 10 but not in association with resurgence. However, one would like to know more about the time course: when does the reignition occur relative to the first spontaneous recurrent seizure? Before it, suggesting that the resurgence of gene expression might cause the first seizure? Or after it (Figure 1B)? Are STAT3 and JAK1 unique, or do other genes that change after SE also exhibit “reignition”? For example, some studies suggest brain-derived neurotrophic factor (BDNF) is elevated when it is studied weeks after SE. 11 Notably, BDNF is thought to be a trigger of the JAK/STAT pathway and alters GABAA receptors, potentially synergizing with effects on STAT3. 12
To their credit, human tissue from patients with TLE was also examined. Although these samples were from patients with chronic epilepsy, not from a time considered to be early during epileptogenesis, analysis of the GRN still showed that STAT3 was a major factor.
Next, the author tried to antagonize STAT3 at the “reignition” phase, so they treated mice chronically with a JAK antagonist. The drug, CP690550 (Tofacitinib), is already Food and Drug Administration-approved and used for autoimmune diseases, cancer, and other conditions. 7 There are some side effects (increased risk of infection, probably due to suppression of the immune system) but the drug seems generally tolerable. 13
Convulsive seizures were reduced in frequency after CP690550 was administered for 2 weeks (compared to vehicle-treated mice). These were very exciting results, but the most intriguing finding might be that the reduction in seizure frequency persisted even after the drug was stopped. In these experiments, they administered CP690550 over 2 weeks. In mice that showed the greatest effect, 80% were seizure-free when they were followed for an additional 4 weeks without drug treatment. These remarkable results are very exciting, and “spark” curiosity: what occurs in the brain to make intractable epilepsy stop requiring treatment? Or would seizures ultimately return, and if so, could CP690550 be used again and still be as effective? These and many other questions will be valuable to advance the promising preclinical results to clinical trials.
Importantly, the authors also addressed comorbidities, a significant concern for people with epilepsy. The authors used two behavioral tasks to assess memory, a common comorbidity in TLE. Although similar, the tasks test different aspects of memory. Mice treated with CP690550 were improved compared to vehicle-treated mice. Therefore, CP690550 not only improved seizures but also two tasks that test memory. The study also included an analysis of markers of gliosis and microglia. Remarkably, CP690550 treatment reduced the typically high level of these markers in epileptic mice. Moreover, for those markers that were studied after treatment stopped, there was evidence of a persistent reduction. Thus, the authors showed that treatment with CP690550 could reduce chronic seizures, comorbid behavioral impairment, and inflammatory markers. Moreover, many effects persisted after drug treatment. Additional studies were also included, many of which added further support and depth to their work. The authors should therefore be highly STAT-isfied. Hopefully, this type of research will “ignite” many others and “fuel” new treatments for patients with epilepsy.