
Abstract
Nasrallah K, Berthoux C, Hashimotodani Y, Chávez AE, Gulfo MC, Luján R, Castillo PE. Cell Rep. 2024; 43(7):114382. doi: 10.1016/j.celrep.2024.114382. Retrograde signaling at the synapse is a fundamental way by which neurons communicate and neuronal circuit function is fine-tuned upon activity. While long-term changes in neurotransmitter release commonly rely on retrograde signaling, the mechanisms remain poorly understood. Here, we identified adenosine/A2A receptor (A2AR) as a retrograde signaling pathway underlying presynaptic long-term potentiation (LTP) at a hippocampal excitatory circuit critically involved in memory and epilepsy. Transient burst activity of a single dentate granule cell induced LTP of mossy cell synaptic inputs, a BDNF/TrkB-dependent form of plasticity that facilitates seizures. Postsynaptic TrkB activation released adenosine from granule cells, uncovering a non-conventional BDNF/TrkB signaling mechanism. Moreover, presynaptic A2ARs were necessary and sufficient for LTP. Lastly, seizure induction released adenosine in a TrkB-dependent manner, while removing A2ARs or TrkB from the dentate gyrus had anti-convulsant effects. By mediating presynaptic LTP, adenosine/A2AR retrograde signaling may modulate dentate gyrus-dependent learning and promote epileptic activity.
Commentary
One of the targets for epilepsy treatment is adenosine. 1 Thus, raising adenosine levels by inhibiting the major enzyme responsible for its metabolism, adenosine kinase, inhibits induced seizures in normal mice and chronic seizures in epileptic mice. 1
Adenosine acts on Purine type 1 (P1) receptors (A1, A2A, A2B A3) which are either inhibitory (A1, A3) or excitatory (A2A, A2B) G proteins (Gi/o; Gs respectively). After it was shown that adenosine rises after seizures in humans with temporal lobe epilepsy (TLE), it was suggested that adenosine might be an endogenous anticonvulsant. 2 Additional experiments in rodents then showed that adenosine actions at A1Rs are anticonvulsant but A2ARs may not be. 1 Although some studies support inhibitory effects of A2ARs on seizures 3 other studies show A2ARs promote seizures. 4
In this context it is interesting that Nasrallah et al 5 describe numerous elegant experiments where they show A2ARs may be proconvulsant. They focus on retrograde release of adenosine from dentate gyrus granule cells (GCs) to presynaptic terminals of mossy cells, a glutamatergic cell of the dentate gyrus hilus. The focus on GCs is relevant to epilepsy since GCs have been implicated repeatedly as a control point or “gate” for seizures. Retrograde signaling is also important to address because it has been implicated in long-term potentiation (LTP), and many researchers consider mechanisms of long-term plasticity to be shared with processes underlying epileptogenesis. However, retrograde signaling has been relatively unexplored in epilepsy.
The synapses of mossy cells on GCs are also important in epilepsy because mossy cells can either promote GC excitation or indirectly inhibit them by activating local GABAergic neurons (Figure 1A). In TLE, there is extensive mossy cell loss. It was initially suggested that mossy cell loss leads to epilepsy because there is less GABAergic inhibition of GCs. However, the excitatory input of mossy cells to GCs appears to be proconvulsant during the initial insult that initiates epileptogenesis. 6
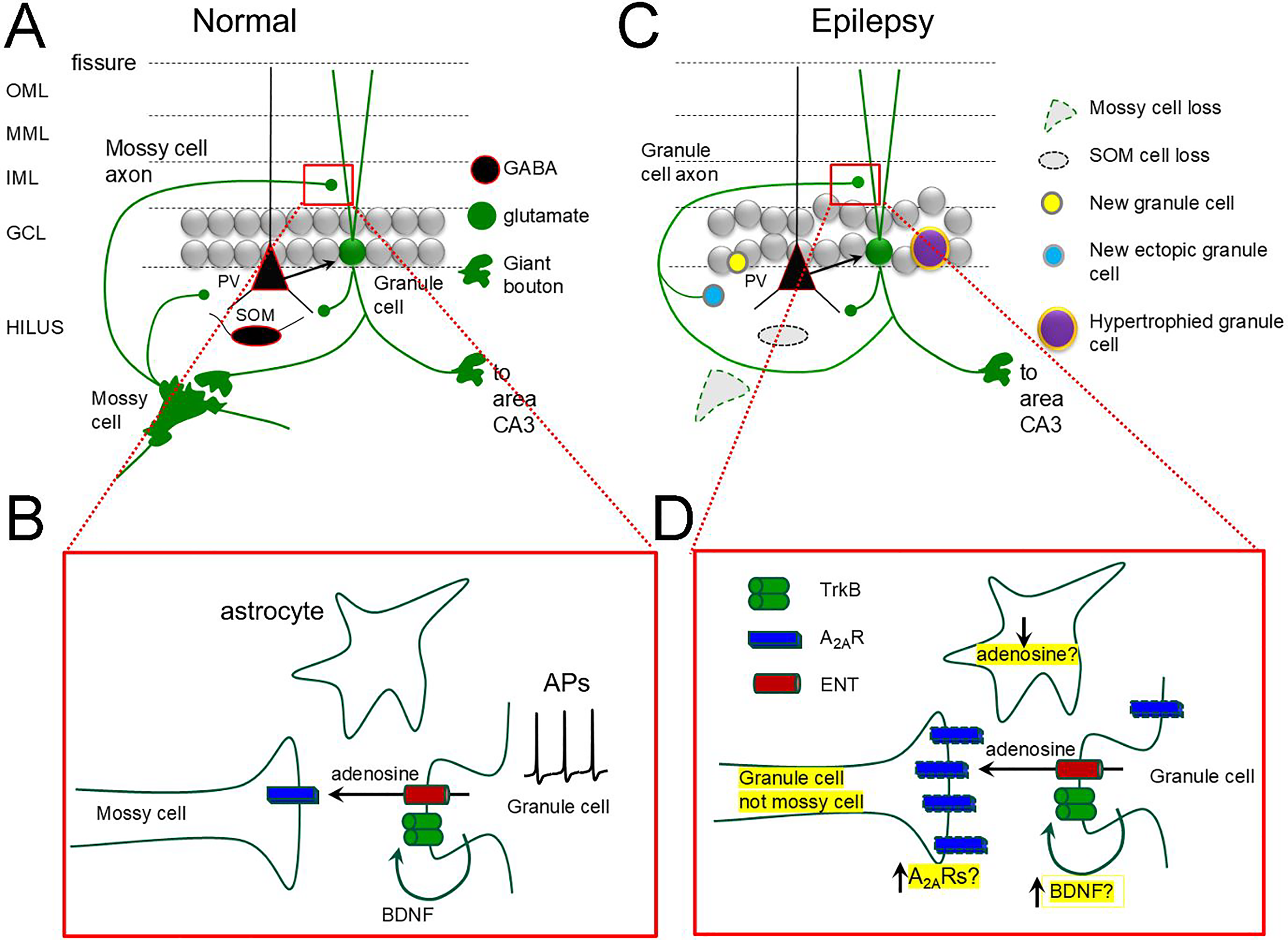
A A simplified schematic of the dentate gyrus cell types and circuitry. A compact layer of glutamatergic granule cells is surrounded by a molecular layer with outer, middle and inner subdivisions where their dendrites are located, and a polymorphic layer (hilus) containing glutamatergic mossy cells and diverse GABAergic neurons (eg, parvalbumin-expressing, PV and somatostatin-expressing, SOM). Granule cells have giant boutons that innervate mossy cells and CA3 pyramidal cells and conventional boutons on GABAergic neurons. Mossy cells innervate GCs and GABAergic neurons.
Nasrallah et al 5 first studied normal mice, so all mossy cells were present. The main question was based on prior studies of the authors showing that stimulating GCs in bursts at theta frequency could evoke LTP of the MC→GC synapse which was presynaptic in mechanism. They showed GC release of BDNF was triggered by GC stimulation, and BDNF acted on one of its receptors, TrkB, located on GC dendrites. TrkB activation then led to release of a retrograde signal to induce LTP. The present study tried to identify the retrograde signal. The authors used pharmacology and clever combinations of transgenic mice and adeno-associated viruses (AAVs) with patch clamp recordings in hippocampal slices to show the retrograde signal was adenosine. Furthermore, adenosine was released passively by a mechanism known to occur elsewhere: an equilabrative nucleoside transporter (ENT). Once adenosine was present extracellularly it bound to (A2ARs) on mossy cell terminals. To their credit, the authors confirmed with electron microscopy that mossy cell terminals contain A2ARs. Thus, their data suggested that adenosine promotes excitation of GCs by MCs after bursts of MC activity (Figure 1B). Theoretically, GC excitation might be decreased by increased activation of GABAergic neurons by mossy cells, but the authors suggested that their presynaptic LTP does not occur at the MC→GABAergic neuron synapses. Thus, the data suggested that adenosine would promote, not inhibit, excitation of GCs, and potentially exacerbate seizures.
To address seizures more specifically they used systemic KA injection to induce seizures and an in vivo sensor of adenosine to show that acute seizures released adenosine, as occurrs in human TLE (described above). In addition, they showed adenosine release was dependent on TrkB, although whether it was GC TrkB specifically was not established. However, they removed TrkB from the glutamatergic neurons of the hippocampus and found anti-convulsant effects, consistent with prior studies (McNamara and Scharfman, 2012). Importantly, the authors showed that deletion of A2ARs in hippocampal glutamatergic neurons had similar effects.
Thus, the authors built a fascinating story starting with the description of a presynaptic form of LTP in normal animals in vitro and then showing its potential relevance to seizures in vivo. It should be emphasized how many difficult and time-consuming approaches were taken.
Now one must grapple with the conundrum that adenosine is typically anticonvulsant but the authors show adenosine is also proconvulsant. How can these opposing effects be reconciled? The explanation that inhibitory effects occur via A1Rs and excitatory effects by A2Rs does not explain why the inhibitory effects are usually dominant. One also wonders how much of what the authors have found occurs in the setting of mesial temporal sclerosis (MTS) in TLE. After MTS, mossy cell loss occurs, and the inner molecular becomes innervated by new axon collaterals of GCs (Figure 1C). How much of what the authors described for the MC→GC synapses also occurs for the GC→GC synapse is relevant to TLE. A2ARs increase in the terminals of glutamatergic neurons in TLE and in astrocytes 7 (Figure 1D). Activated astrocytes have low levels of adenosine due to increased metabolism by adenosine kinase (Figure 1D) and the authors did not discuss the role of astrocytes. It is likely to be important because correcting the low adenosine levels by inhibiting adenosine kinase shows remarkable efficacy in preclinical models. 1
Other questions also remain to be answered. Is the presynaptic plasticity the same for adult-born GCs? These GCs have special roles compared to the mature GCs, heightened plasticity, and influence seizures and chronic epilepsy. 8 Mossy cells have a special relationship to young GCs born in the adult brain, forming one of their first excitatory inputs in the inner molecular layer. Therefore, in light of the data from Nasrallah et al, 5 mossy cells would be in a powerful position to induce robust LTP in young GCs. Mossy cells also innervate ectopic GCs, which have been implicated in epilepsy. 9 If the findings of the authors about mossy cell synapses on GCs in the cell layer also pertain to mossy cell synapses on ectopic GCs, that would promote seizures also.
Although the study leads to many questions, the paper provides important insights into long-standing questions about the GCs and epilepsy. With respect to BDNF, GCs have some of the highest levels in the CNS normally, and BDNF increases further in chronic epilepsy, both in rodents and humans (Figure 1C). However, assays of BDNF have shown it either is transported to GC dendrites or GC axons, the mossy fibers.10,11 Most research to date has focused on mossy fiber BDNF.12) But what about dendritic BDNF? The studies of Nasrallah et al suggest that, like the GC terminals, release of BDNF from GC dendrites binds locally to TrkB to promote GC excitation, and potentially seizures. This may be one reason why TrkB inhibition in GCs is so effective in hippocampal seizures. 12
Hopefully these and other valuable ideas from Nasrallah and colleagues will make more investigators start no-tic-ing aden-o-sine.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (grant numbers R37 NS-126529, R01 AG-055328) and the New York State Office of Mental Health.