
Abstract
Cuhadar U, Calzado-Reyes L, Pascual-Caro C, Aberra AS, Ritzau-Jost A, Aggarwal A, Ibata K, Podgorski K, Yuzaki M, Geis C, Hallerman S, Hoppa MB, de Juan-Sanz J. Cell Rep. 2024;43(5):114186. doi:10.1016/j.celrep.2024.114186. The fine control of synaptic function requires robust trans-synaptic molecular interactions. However, it remains poorly understood how trans-synaptic bridges change to reflect the functional states of the synapse. Here, we develop optical tools to visualize in firing synapses the molecular behavior of two trans-synaptic proteins, LGI1 and ADAM23, and find that neuronal activity acutely rearranges their abundance at the synaptic cleft. Surprisingly, synaptic LGI1 is primarily not secreted, as described elsewhere, but exo- and endocytosed through its interaction with ADAM23. Activity-driven translocation of LGI1 facilitates the formation of trans-synaptic connections proportionally to the history of activity of the synapse, adjusting excitatory transmission to synaptic firing rates. Accordingly, we find that patient-derived autoantibodies against LGI1 reduce its surface fraction and cause increased glutamate release. Our findings suggest that LGI1 abundance at the synaptic cleft can be acutely remodeled and serves as a critical control point for synaptic function.
Commentary
Proper synaptic activity requires the coordinated activity of numerous pre- and postsynaptic proteins and their coupling via trans-synaptic molecules. These molecules regulate synapse formation and morphology, receptor function, synapse elimination, and dynamic neuroplasticity driven by neural activity. Numerous disease processes, including epilepsies, are related to mutations in genes that regulate various aspects of synaptic function, the so-called synaptopathies.
A neuronal glycoprotein called LGI1 (leucine-rich glioma inactivated 1) is part of a complex that regulates synaptic function and excitability.1,2 LGI1 is essential for circuit development and function. 3 It modulates neuronal excitability through its action on presynaptic voltage-gated potassium Kv1.1 channels and modulates glutamatergic synaptic transmission by regulating the surface expression of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptors. LGI1 dysfunction underlies two well-recognized epilepsy etiologies – genetic and autoimmune. Genetic mutation of LGI1 causes autosomal dominant lateral temporal lobe epilepsy (ADLTE) while acquired antibodies to LGI1 lead to LGI1-limbic encephalitis (LE).
ADLTE, originally called autosomal dominant partial epilepsy with auditory features, 4 involves inherited mutations in LGI1 and occurs across the age spectrum. Seizures are often auditory in nature (focal aware) and can spread bilaterally. EEG shows interictal temporal lobe discharges and MRI scans are usually normal. Treatment relies on standard antiseizure medications.
Autoimmune LE is caused by antibodies to LGI1 and is most often seen in elderly males. 5 Seizure foci are located in the medial temporal lobe and/or motor cortex. Encephalitic symptoms such as cognitive impairment and memory dysfunction relate to limbic system dysfunction and often have a gradual, subacute onset. Neurocognitive symptoms may be preceded by a unique seizure semiology (considered pathognomonic of LGI1-LE) called fascio-brachial dystonic seizures, the occurrence of which implies involvement of motor cortex as well as the limbic system. 6 MRI scans show limbic hyperintensities and immunotherapy is the most effective treatment.
Thus, dysfunction of LGI1 can underlie a wide spectrum of human epilepsy and has been validated in LGI1 knockout mice. 7 Full understanding the clinical features and potential treatments for these disorders requires a detailed clarification of the synaptic pathophysiology. The mechanisms by which LGI1 mutations or antibodies lead to epilepsy are largely unknown. Unraveling the molecular details of LGI1 function therefore represents a prime example of how enhanced understanding of cellular function can inform clinical care. The advances reported here by Cuhadar and colleagues were largely facilitated by the development of novel optical tools (fluorescent labels) that permit monitoring of LGI1-associated molecules within a neuron. 8 One fluorophore label, LGI1-pHluorin (LGI1-pH), allowed the tracking of LGI1 movement and translocalization within presynaptic arborizations of single neurons as a function of neuronal firing, utilizing primary co-cultures of rat hippocampal neurons and astrocytes. Comprehending the findings requires an appreciation of multiple interacting molecular components (Figure 1A).
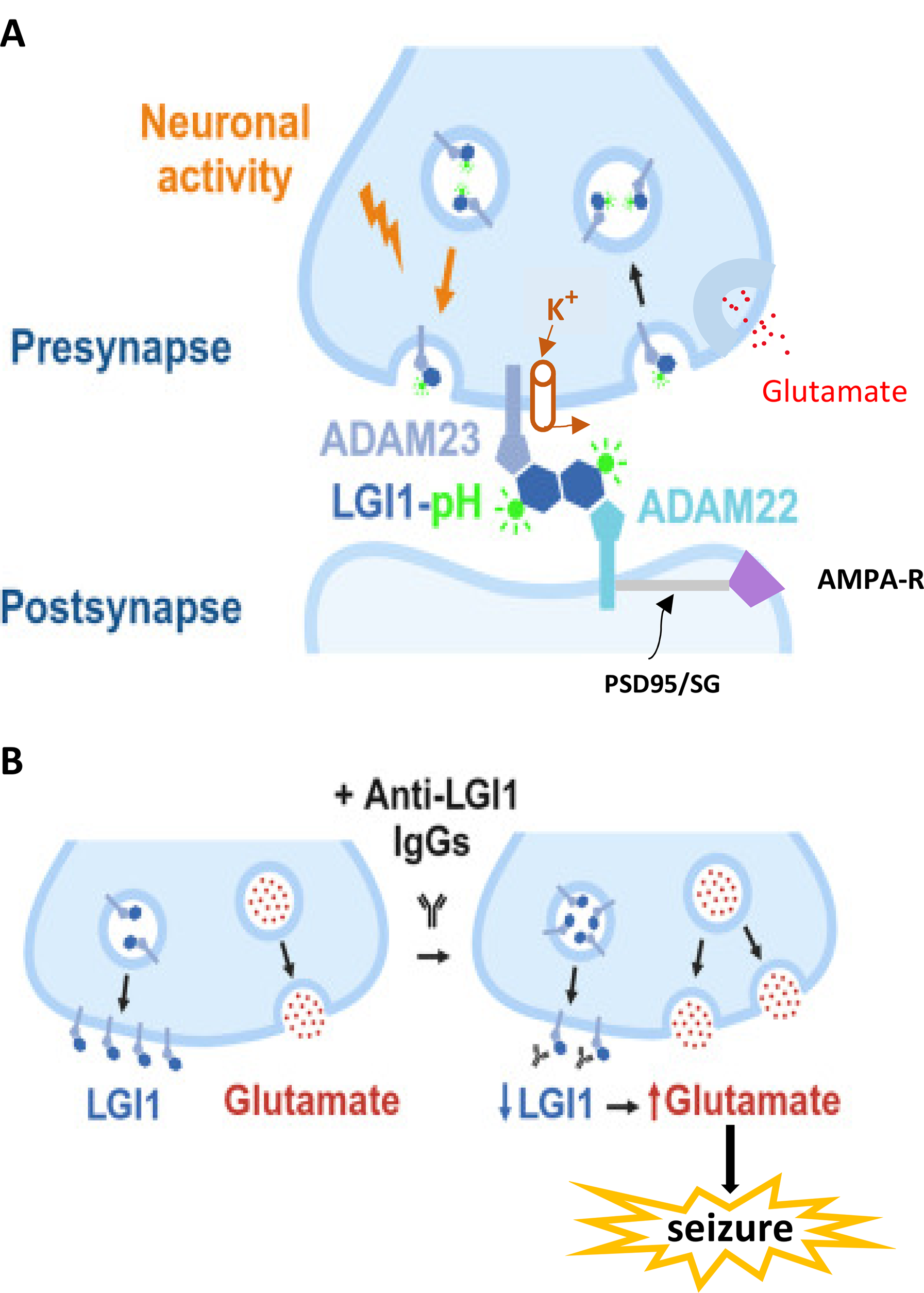
Molecular architecture of a synapse and the role of LGI1 binding partners in normal and pathological conditions. (A) Normal presynaptic and postsynaptic components are shown. Presynaptically, neuronal firing causes the translocation of LGI1 bound to ADAM23 to the presynaptic cleft surface. (Within the cleft, binding partners are shown in enlarged form.) The green molecule attached to LGI1 is a fluorescent tag, pHluorin (LGI1-pH), that permits tracking of movement and abundance of the complex. Kv1.1 potassium (K+) channels are linked to the LGI1/ADAM23 complex, providing indirect control of presynaptic excitability by stabilizing membrane potential and keeping action potential duration short, thereby constraining glutamate release. After exocytosis, LGI1/ADAM23 complexes can be endocytosed back into the presynaptic terminal (black arrow on right). Postsynaptically, LGI is linked to ADAM22. This complex modulates the expression of AMPA-type glutamate receptors (purple trapezoid) that are linked to ADAM22 by postsynaptic density (PSD) 95 and the Stargazin (SG) complex (gray bar). Mutations in LGI1 could reduce K+ channel activity and favor increased excitability and seizures. The role of decreased AMPA receptor expression in LGI1-related seizure genesis is uncertain. (B) Examples of normal (left) and pathologic (right) LGI1 function. At baseline (left), an abundance of LGI1/ADAM23 permits modest glutamate release, related to the high expression of LGI1 via its effects on Kv1.1 channels. When the synapse is exposed to antibodies to LGI1 (Y-shapes, right), LGI1 expression is decreased, leading to increased glutamate release and possibly generating seizure activity. Reproduced and modified from Ref. 8 with permission from Elsevier/Cell Press.
Prior to this work, it was known that LGI1 binds to one of two receptors, ADAM23 or ADAM22 (
Presynaptic Kv1.1 potassium channels are linked to ADAM23/LGI1, leading to stabilization of membrane potential and prevention of excessive neurotransmitter (glutamate) release from the presynaptic terminal. The amount of LGI1 present at the synaptic cleft constrains glutamate release because K
This double dimer (LGI1/ADAM23 and LGI1/ADAM22) regulates both presynaptic neuronal excitability and postsynaptic AMPA receptor expression. Therefore, LGI1 regulates aspects of synaptic function that are pivotal for the emergence of seizure activity. Dysfunction of LGI1 or its partners would be pro-epileptic at excitatory synapses due to hyperexcitability induced by reduction of potassium currents and consequent increased glutamate release. Postsynaptically, LGI1/ADAM 22 regulates AMPA receptor expression at the membrane surface. Defective LGI1 function would alter AMPA receptor presence at the postsynaptic surface though it is unclear how this would affect excitability.
The work by Cuhadar et al adds to the prior knowledge of LGI1 function in several ways. Numerous mutations in LGI1 have been identified with variable clinical consequences. The kinetics of translocation of the ADAM-LGI1 complex to the surface membrane and strength of binding differ according to the mutation site (more than 45 of which have been identified). One goal of the study of Cuhadar and colleagues was to use novel optical tags on LGI1 to follow its route through the presynaptic terminal to the presynaptic membrane surface. These techniques required the determination of pH of the various subcellular compartments that affect the optical fluorescence changes. Likewise, postsynaptic fluorescence changes were utilized to characterize the subcomponents of the synaptic dysfunction. Altogether, the authors showed that presynaptic neuronal activity drives LGI1 translocation to the synaptic surface, where its abundance reflects neuronal firing activity. LGI1 is not localized within synaptic vesicles and then secreted (as previously thought), but rather traffics along with ADAM23 to be exocytosed and then endocytosed back into the presynaptic terminal. They showed that the abundance of LGI1 at the synaptic cleft surface controls presynaptic action potential waveform, subsequent presynaptic calcium entry, synaptic vesicle cycling, and glutamate release. The spectrum of epileptic LGI1 mutations reflects different defects in its synaptic translocation or stabilization. Finally, exposure of the neuron cultures to antibodies from patients with LGI1-LE showed that the antibodies decreased surface LGI1 abundance and increased glutamate release, consistent with increased seizure predisposition (Figure 1B).
These experiments are a tour-de-force in subcellular characterization of a set of molecules intimately involved in synaptic regulation, dysfunction of which leads to seizures in two broad classes of LGI1 dysfunction. The authors assert that the techniques could lead to development of an assay system that would be useful clinically, to define various mutation-phenotype correlations. Given that the occurrence of LGI1 dysfunction in humans is not trivial (LGI1-LE is second only to N-methyl-
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.