
Abstract
Dendrites are tree-like structures with tiny spines specialized to receive excitatory synaptic transmission. Spino-dendritic plasticity, driven by neural activity, underlies the maintenance of neuronal connections crucial for proper circuit function. Abnormalities in dendritic morphology are frequently seen in epilepsy. However, the exact etiology or functional implications are not yet known. Therefore, to better comprehend the structure-function significance of this dendritic pathology in epilepsy, it is necessary to identify the common spino-dendritic disturbances present in both human and experimental models. Here, we describe the dendritic and spine structural profiles found across human refractory epilepsy as well as in animal models of developmental, acquired, and genetic epilepsies.
Introduction
Dendrites are tree-like extensions that are specialized to receive information transmitted between neurons. Dendrites have microscopic protrusions called dendritic spines, which are sites for excitatory synaptic transmission. Dendritic spine shapes are highly dynamic and can change in response to neural activity driven by behavioral and cognitive experiences. 1 For example, stimuli driving learning and memory processes can result in the remodeling of existing spines as well as the generation of new spines (spinogenesis). This synaptic plasticity is thought to underlie the maintenance of neuronal connections necessary for cognitive development. As such, neuronal function and cognitive behavior can be deeply affected by subtle changes in dendritic and spine morphology. Abnormalities in dendritic architectures are often found in epilepsy 2,3 and neurological disorders like Alzheimer’s disease, 4 which are characterized by cognitive decline. However, the exact causes and functional implications of the dendritic pathology in epilepsy are not definitively known. 2,3 It is well established that dendritic structure and function are strongly coupled. This indicates that an increase in spine density and/or the presence of mushroom-like or stubby-shaped spines may result in greater excitatory input compared to thin spines. Consequently, the functional impact of changing spine density may vary depending on which type of spine is affected. Therefore, the identification of spino-dendritic structural pathology across human and animal models of epilepsy can help us better understand the potential functional impact. Here we describe and summarize the spino-dendritic structural profiles found in human refractory epilepsy as well as in animal models of developmental, acquired, and genetic epilepsies.
Dendritic Profiles in Human Epilepsy
Since the first descriptions of the phenomenon of blebbing or swollen dendritic spines in brains from individuals with epilepsy, observed by DeMoor in 1898, neurologists and scientists have been intrigued by the presence of these “unique” dendritic morphological abnormalities in epileptic illnesses (reviewed in the study by Swann et al 5 ). Then after, numerous investigations have consistently found areas of focal dendritic swelling known as dendritic beading along with sparse dendritic spines and simplified dendritic architectures across different brain regions that include the neocortex, hippocampus, and amygdala derived from refractory epilepsy cases (e.g., temporal lobe epilepsy [TLE] and focal cortical dysplasia [FCD]). 5 -10
Dendritic structures in neocortical and hippocampal pyramidal neurons from TLE patients often exhibit reduced branching, the presence of bead-like shafts, and loss of dendritic spines. 6,11 The appearance of dendritic beading, characterized by abnormal patterns of microtubule whirling, was specifically localized in apical dendrites. 7 Within amygdala neurons from TLE cases, the dendritic structures showed reduced numbers of first-order branches, with the remaining dendrites showing nodular varicosities, dendritic shaft swelling, spine bifurcation, and pronounced constrictions. 9 Furthermore, a comparison of dendritic architectures in FCD cases such as FCDIIa and FCDIIb, 6 which are characterized by the presence of dysmorphic neurons, 12 revealed contrasting changes in dendritic complexity in normal looking neurons and dysmorphic neurons. Normal looking neurons showed decreases in dendritic branching and spine density, while dysmorphic neurons showed increased dendritic complexity and more filopodia-like protrusions. 6 It is possible that dysmorphic neurons may carry mutations within the mechanistic target of rapamycin (mTOR) pathway, which may directly influence their dendritic structure, while nearby seemingly normal neurons may show secondary effects in their dendritic arbors. Further, the areas of reduced spine densities in type II FCD cases correlated with increased immunoreactivity for microglial markers and complement proteins C1q and C3. 8 These findings, along with evidence of colocalization of C1q to Map2-labeled dendrites in areas with fewer dendrites in FCD 13 suggest that complement-mediated neuroimmune interactions may contribute to dendritic structural abnormalities in epilepsy (for review see the study by Wyatt-Johnson and Brewster 14 ).
The clear absence of dendritic structural abnormalities in brains of autopsy controls 5 -9,11 raises the possibility that the aforementioned dendritic structural pathology is unique to epilepsy. Indeed, the observation that neocortical areas with high interictal spiking exhibited reductions in both the density and length of silver-stained fibers in layer IV neurons compared to areas of low spiking activity in the human epileptic brain 10 suggests a potential role for network hyperactivity in the destabilization of dendritic structures that occurs in human epilepsy.
Dendritic Profiles in Experimental Models
Early Life Seizures
During prenatal and postnatal development, dendrites grow (dendritogenesis), spines emerge, and new synapses form (synaptogenesis). Dendritic growth can continue until adulthood (reviewed in the study by Prigge and Kay 15 ), while synaptogenesis peaks in the early postnatal period (reviewed in study by Sudhof 16 ), but also continues in adulthood. Subsequently, pruning of dendrites, spines, and synaptic structures plays a crucial role in shaping functional neural connectivity. 1,15,16 Dendritogenesis and pruning are carefully orchestrated by a combination of genetic programs and neural activity. 1,15,16 However, events such as early-life seizures (ELS) can disrupt these processes and harm the proper network connectivity and function of the developing brain. 17
Experimental models of ELS occurring between the first and second weeks of postnatal development are associated with long-lasting alterations of hippocampal dendritic architectures 18 -22 and hippocampal-dependent spatial learning and memory dysfunctions. 17,18,20,21 Dendritic structural alterations in CA1 pyramidal cells and dentate granule cells (GCs) have been consistently reported in studies utilizing different models of ELS induced by the volatile convulsant flurothyl, 20 hyperthermia, 18,22 chemoconvulsants, 21 or hypoxia. 19 Seizures triggered with flurothyl inhalation during postnatal days (P) 7 to 11 in mice resulted in decreased basal dendrite length and branching of CA1 neurons and impaired spatial learning and memory functions 6 weeks later. 20 The induction of experimental febrile status epilepticus (SE) through hyperthermia in P10 rats was linked to a reduction in the length of CA1 apical dendrites and an increase in the complexity of GC dendritic structures, which correlated with reduced spatial memory functions 12 weeks after the ELS. 18 Because it is well established that seizures can promote neurogenesis of GCs that integrate into existing GC circuitry, 23 a different study evaluated the dendritic and spine morphology of newborn GCs between 1- and 8-weeks post febrile seizures in P10 rats. 22 This study found that newborn GCs exhibited longer dendrites with increased spine densities, specifically within the molecular layer of the dentate gyrus. 22 In line with these findings, a time course analysis performed between 1 and 3 weeks after ELS induced with the chemoconvulsant kainic acid (KA) in P14 mice revealed an initial rise in GC dendritic branching and spine density along with a persistent reduction in dendritic branching within CA1 and CA3 cells. 21 Additionally, an increased abundance of thin-shaped spines coupled with a decreased density of stubby-shape spines in CA1 dendrites was observed between 1- and 4-weeks following hypoxia-induced seizures in P10 rats 19 as well as following KA in immature mice. 21 Given that short CA1 dendrites, arborized GC dendrites, and thin-shaped spines are indicative of an immature state, 17 it is possible that ELS may disrupt the maturation of dendritic architectures.
These studies agree that ELS is associated with dendritic complexity that is decreased in CA1 and increased in GCs. Underlying mechanisms may be related to impaired growth or excessive pruning of CA1 dendrites or amplified growth or inadequate pruning of GCs, potentially involving signaling cascades such as neuron-restrictive silencer factor (NRSF), 18 calcineurin, 24 immune complement molecules, 8,14 microglia, 21 and the mTOR pathway (discussed next). It is noteworthy that treatments involving minocycline 21 and NRSF signaling inhibitors 18 attenuated some of the dendritic pathology and improved hippocampal-dependent functions. This finding suggests that ELS may contribute to the development of behavioral and/or cognitive impairments later in life.
Acquired Epilepsy
Brief seizures and episodes of SE in adults can cause immediate and enduring alterations in the morphology of dendritic arbors within the neocortex and hippocampus. Evidence of rapid seizure-induced dendritic changes comes from in vivo timelapse multiphoton imaging of neocortical neurons in adult mice, which revealed that electrographic seizures triggered with 4-aminopyridine (4AP) or KA resulted in the immediate development of dendritic beading and spine loss. 25 -28 This effect was aggravated with increased seizure severity 25 -28 and was facilitated by the disruption of the actin cytoskeleton through calcineurin-dependent signaling. 25 In addition, a single generalized seizure induced in adult rats using the chemoconvulsant pentylenetetrazol was sufficient to cause a mTOR-dependent loss of CA1 dendritic spines within 3 hours. 29 These findings support the idea that spino-dendritic structures are susceptible to rapid plastic changes in response to seizure activity.
Longer periods of seizure activity associated with SE events can lead to long term severe loss in spine and dendritic density in the neocortex and hippocampus, along with the subsequent development of spontaneous recurrent seizures (SRS) and cognitive decline. 26,28,30 -32 Within the neocortex, >50% of spines were lost between 24 hours and 6 weeks after KA. 26,28 Within hippocampal CA1 dendrites, a 15% to 20% loss in spine density along with a 30% decrease in dendritic branching were observed during the period of epileptogenesis between 1 and 3 weeks after SE. 30 -32 Status epilepticus-induced decreases in dendritic branching and spine density within CA1 and the neocortex were prevented or mitigated by inhibiting the mTOR pathway. 26,30 This effect corresponded with enhanced learning and memory abilities in rats that experienced SE. 30 In contrast to these observations in CA1, significant increases in CA3 spine numbers (∼30%) 33 and increases in GC dendritic branching and spine density 34 were reported during the chronic epilepsy phase following SE. A recent review by Jean et al 35 further describes differences in the dendritic spine morphology within the hippocampus during epileptogenesis and discusses potential underlying mechanisms. Taken together, these findings support an association between SE and enduring alterations in dendritic structures. However, despite this compelling evidence, a definitive link between dendritic structural abnormalities and the subsequent development of SRS and cognitive decline remains elusive.
Genetic Epilepsy
Due to the essential role the mTOR pathway plays in modulating neuronal and dendritic structures under physiological and pathological conditions, including epilepsy, 36,37 numerous studies have been conducted to determine how genetically enhancing mTOR activity can lead to dendritic structural alterations and trigger epilepsy. 38 -40 For example, genetic ablation of the upstream mTOR regulatory molecule the phosphatase and tensin homolog (Pten) renders mTOR constitutively active and promotes epilepsy. 37 Pten deletion in hippocampal dentate GCs results in dendric overgrowth, neuronal hyperexcitability, and epilepsy that resembles human FCD. 38 Granule cells that lack Pten exhibit complex dendritic branches with an augmented number of spines and increased excitability. 38,41 -43 Some mechanisms implicated in the extensive dendritic structures observed in Pten-negative GCs include an increase in the rate of microtubule polymerization 44 and downstream signaling of the mTOR complex 2. 42 Similar dendritic overgrowth occurs in association with loss-of-function mutations in TSC 45 and DEPDC5 molecules, 46,47 which negatively regulate mTOR signaling and also result in epilepsy (reviewed in the study by Jozwiak et al 48 and Samanta 49 ). Although these findings collectively highlight a crucial role for mTOR signaling in the dendritic structural pathology associated with neuronal hyperexcitability, seizures, and epilepsy, a recent study showed that normalization of dendritic structures did not alter seizure severity in Pten null mice. 42 Thus, the precise function of irregular dendritic architectures in the generation of epileptic circuits requires further investigation.
Conclusion
The most common spino-dendritic disturbances found across both human epilepsy cases and experimental models include a reduction in the complexity of dendritic arbors and spine numbers within pyramidal cells of the neocortex and hippocampus (Figure 1A-B). 5 -11,18,20,21,25 -35 Additionally, increases in dendritic branching and spine density are evident specifically in dysmorphic cells in human FCD cases 6,8 and in GCs following ELS 18,21,22 or genetic-induced decreases of mTOR regulatory molecules like Pten, TSC, and DEPDC5 (Figure 1C-D). 37 -42,45 -47 Notably, the presence of dendritic beading seems to be a unique feature related to seizures and epilepsy. These alterations in spines and dendrites likely involve different mechanisms, including pro-epileptogenic modifications (such as those seen in mTOR mutants), adaptive homeostatic adjustments (potentially involving spine loss), and changes due to cellular injury. The stage of the disease can further impact dendritic complexity, as observed in human studies focusing on chronic and refractory epilepsy, in contrast to animal models, which often deal with acute epilepsy conditions.
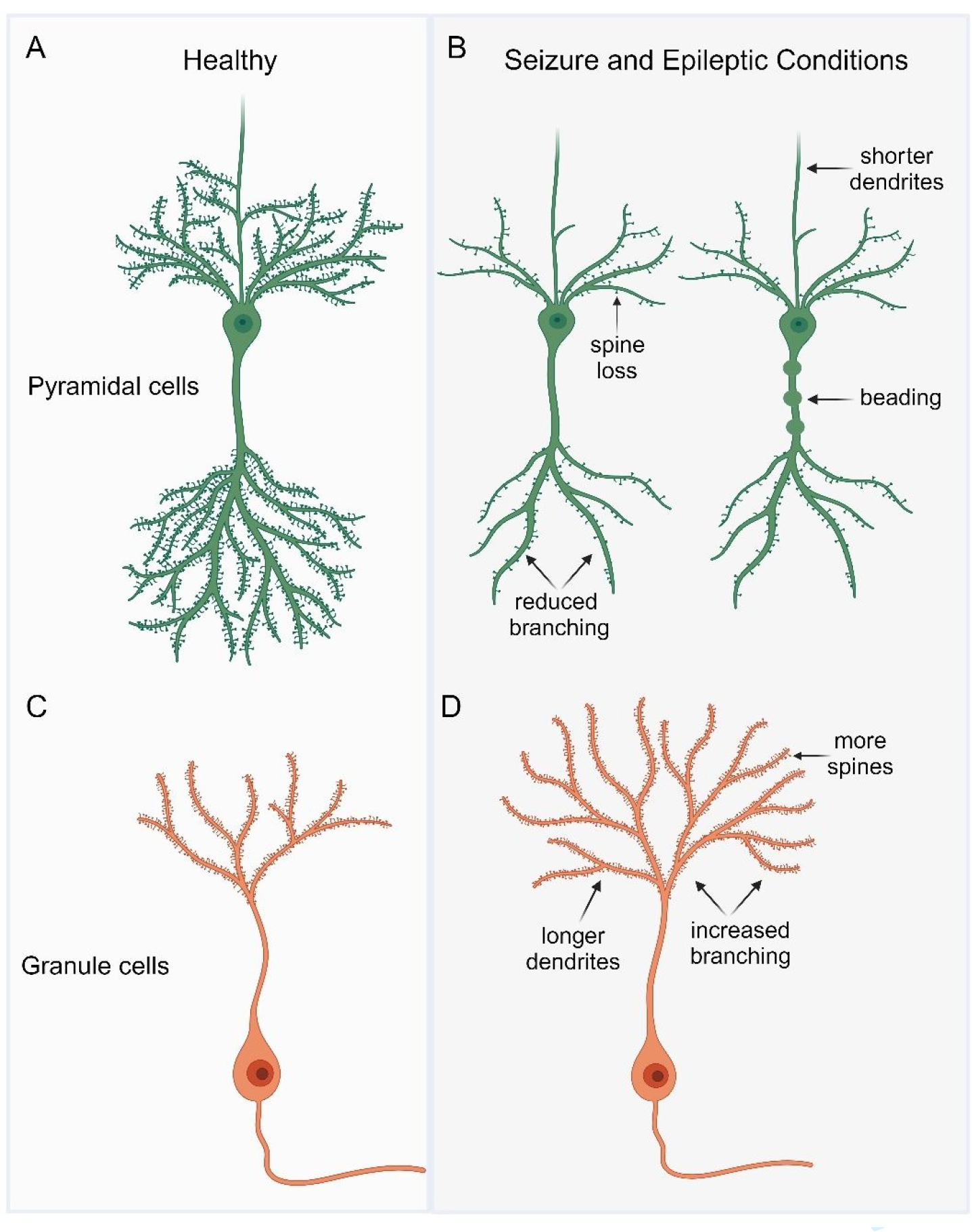
Common spino-dendritic alterations in two primary neuronal types in healthy and epileptic conditions. (A-B) Illustrations of pyramidal neurons under healthy (A) and seizure or epileptic conditions (B). (B) Representative cartoons depicting reduced spine and branching density along with the classic beading phenotype seen in the dendritic apical shaft of pyramidal neurons from seizure or epileptic conditions. (C-D) Illustrations of dentate granule cells under healthy (C) and seizure or epileptic conditions (D). (D) Granule cell cartoon demonstrating increased dendritic complexity including increased spine density and extensive dendritic branching under seizure or epileptic conditions. Created with BioRender.com.
In our view, this evidence suggests a definitive link between dendritic structural abnormalities and epilepsy, though there is still a limited understanding regarding the precise role of these dendritic changes in triggering SRS and cognitive decline. To achieve a comprehensive understanding, we must acknowledge the limitation that the dendritic irregularities may also be connected to inherent methodological challenges, especially in human clinical studies, that include variations in brain tissue acquisition methods such as biopsies for epilepsy cases and autopsies from control cases. Other factors that can impact human dendritic architectures include use of anti-seizure drugs, underlying health conditions, disease stage, age, and sex. In fact, a particular aspect that is notably lacking information relates to sex-differences in dendritic architectures in epilepsy, as only one study discussed here addressed potential sex differences in experimental epilepsy. 43 In human TLE, sex-differences are known to occur at the synaptic level, with men showing higher synaptic density in the temporal neocortex compared to women. 50 Moving forward, this body of evidence from developmental, acquired, and genetic epilepsies can help guide the selection of appropriate animal models and timelines to determine the functional impact of dendritic structural irregularities in experimental epilepsy. Unraveling the processes underlying pathological spino-dendritic remodeling may hold the key to developing innovative therapeutic strategies addressing either the causes or the consequences of alterations in dendritic complexity in epilepsy.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financialsupport for theresearch, authorship, and/or publicationof this article: The funding is provided by National Institute of Neurological Disorders and Stroke (R01 NS096234)