
Abstract
Valassina, N., Brusco, S., Salamone, A, et al. Nat Commun. 2022;13:161. doi:10.1038/s41467-021-27837-w. Dravet syndrome (DS) is a severe epileptic encephalopathy caused primarily by haploinsufficiency of the SCN1A gene. Repetitive seizures can lead to endurable and untreatable neurological deficits. Whether this severe pathology is reversible after symptom onset remains unknown. To address this question, we generated a Scn1a conditional knock-in mouse model (Scn1a
Stop/+) in which Scn1a expression can be re-activated on-demand during the mouse lifetime. Scn1a gene disruption leads to the development of seizures, often associated with sudden unexpected death in epilepsy (SUDEP) and behavioral alterations including hyperactivity, social interaction deficits and cognitive impairment starting from the second/third week of age. However, we showed that Scn1a gene re-activation when symptoms were already manifested (P30) led to a complete rescue of both spontaneous and thermic inducible seizures, marked amelioration of behavioral abnormalities and normalization of hippocampal fast-spiking interneuron firing. We also identified dramatic gene expression alterations, including those associated with astrogliosis in DS mice, that, accordingly, were rescued by Scn1a gene expression normalization at P30. Interestingly, regaining of Nav1.1 physiological level rescued seizures also in adult DS mice (P90) after months of repetitive attacks. Overall, these findings represent a solid proof-of-concept highlighting that disease phenotype reversibility can be achieved when Scn1a gene activity is efficiently reconstituted in brain cells.
Commentary
Dravet syndrome (DS) is a devastating epileptic encephalopathy characterized by frequent seizures, intellectual disability, behavioral abnormalities, developmental delay, sleep disturbance, and other health issues. Seizures in DS are commonly severe, drug-resistant, and sometimes fatal. Seizures in DS also happen early with average seizure onset before six months of age. Tragically, 10–20% of DS patients do not survive beyond ten years of age and nearly half of mortalities are due to sudden unexpected death in epilepsy (SUDEP). 1 There is no cure for DS and the currently available seizure management options are limited and sometimes prove ineffective.
The majority of DS cases are caused by heterozygous loss-of-function mutations in the SCN1A gene which encodes the alpha subunit of the NaV1.1 voltage-gated sodium channel. Hypothetically, restoration of functional SCN1A is a viable approach for treating DS pathologies, including epilepsy. Indeed, genetic rescue studies in mouse models of DS have collectively demonstrated a general proof-of-principle whereby reinstatement of SCN1A can fully or partially correct a wide spectrum of DS pathologies including behavioral abnormalities, development of spontaneous seizures, hyperthermia-seizure susceptibilities, and premature seizure-induced sudden death. 2 These genetic rescue approaches also reversed dysfunctions measured at the circuit, cellular, and molecular levels. Therefore, gene therapy holds great therapeutic promise for treating DS at its source. However, the time window of genetic rescue for therapeutic recovery of DS remains elusive.
In this critical preclinical study, Valassina et al. first developed a novel mouse model of DS harboring a STOP cassette flanked by loxP sites within the intron between exons 6 and 7 of Scn1a. The knock-in mouse (Scn1a Stop/+) enables precise temporal and spatial control of Scn1a reinstatement upon Cre-mediated recombination. Armed with this powerful tool, the authors restored Scn1a expression at both transcript and protein levels during early neonatal (P1–P2) and postnatal (at P30 and P90) periods by systemically delivering a blood-brain barrier permeable AAV-PHP.eB capsid containing the Cre driver. Reinstatement of Scn1a fully rescued seizure and SUDEP phenotypes of DS model mice regardless of the developmental stages when gene reinstatement was initiated. Beyond phenotypic rescue, the authors also took a deep dive into the potential circuit and molecular mechanisms of the genetic rescue of DS. The authors discovered that DS model mice exhibited a global increase of the hippocampal CA1 interneuron excitability after the restoration of Scn1a gene expression, which in turn potentiated the inhibition of nearby pyramidal neurons. The authors also performed differential gene expression profiling and ontology analysis in cortices and hippocampi of Scn1a +/+, Scn1a Stop/+-Ctrl and Scn1a Stop/+-Cre mice to pinpoint the role of astrogliosis in DS manifestations. Whether these circuit and molecular changes are the primary drivers of seizures in DS still requires further investigation.
The majority of developmental and epileptic encephalopathies and many neurodevelopmental disorders with comorbid epilepsy are primarily attributed to pathogenic mutations, deletions, and duplications. Thereby, restoration of the normal level of gene expression provides a promising treatment strategy. A fundamental question that needs to be addressed for guiding the gene therapy clinical trials is “What is the therapeutic window when normalization of gene expression can effectively suppress seizures and other pathologies?” The answers may vary, as animal studies suggest (Figure 1). For example, adult (12 months) reinstatement of Mecp2 can reverse many symptoms including seizures in mouse models of Rett syndrome. 3 Genetic reversal of Syngap1 at P60 in mice improved cerebral function and lowered seizure susceptibility. 4 The therapeutic window for seizure-related phenotypes shifts earlier for Angelman syndrome as juvenile (P21) reinstatement of Ube3a failed to rescue audiogenic seizures in Angelman syndrome model mice. 5,6 P21 treatment did, however, prevent the development of a pro-epileptogenic phenotype when mice were challenged using a flurothyl kindling and retest model. Adult treatment (2-3 months), on the other hand, was not effective with the same challenge. 7 The therapeutic window closes even earlier for seizure rescue in Fragile X mouse models. The delivery of AAV capsid that contained a major isoform of fragile X mental retardation protein at the neonatal stage (P5) failed to halt audiogenic seizures. 8 Of note, besides timing, many other factors including the efficiency and distribution of transgene expression as well as seizure model selection also need to be taken into account when evaluating seizure rescue efficacy via gene normalization approaches.
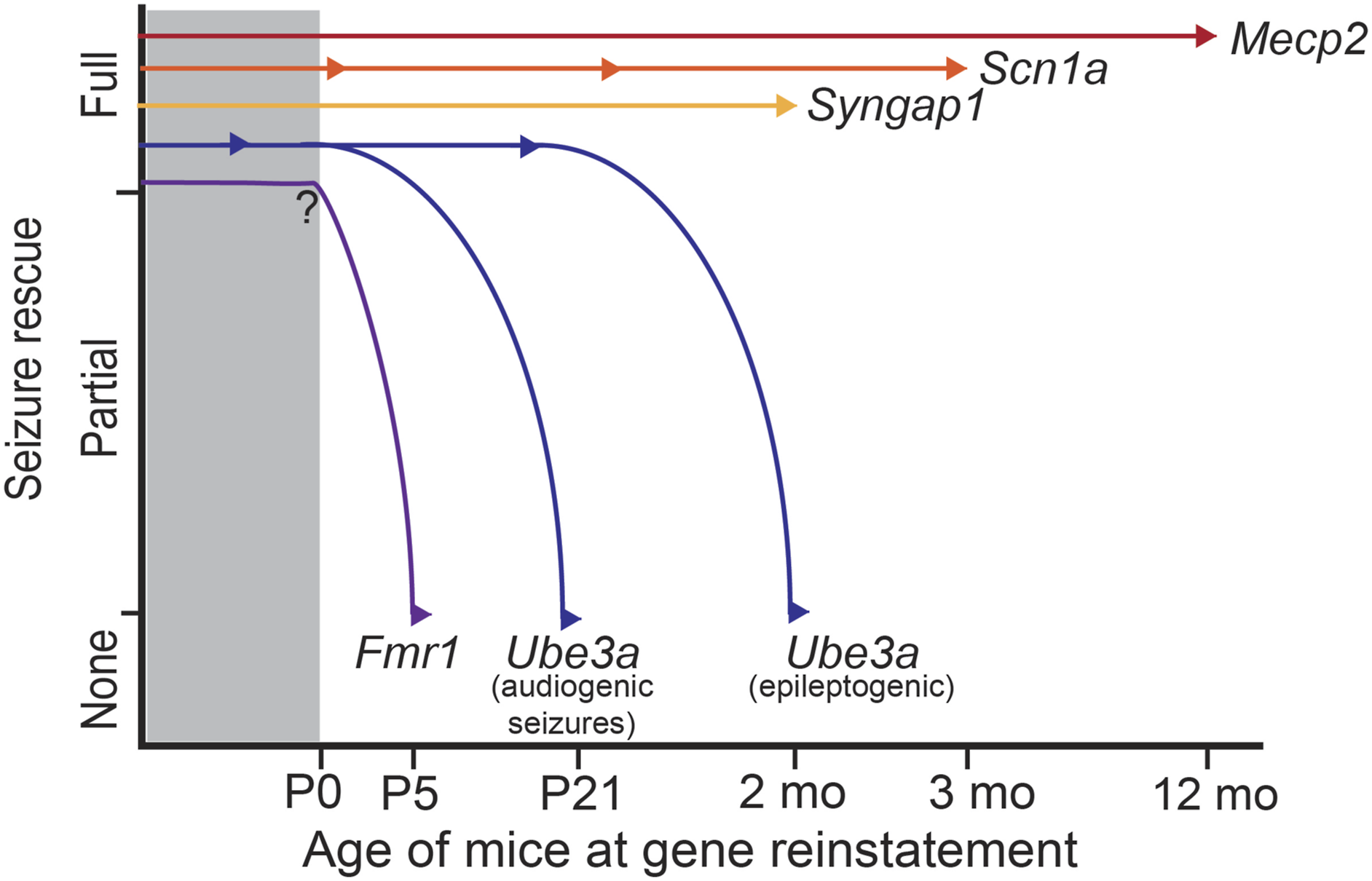
Schematic of seizure rescue efficacy using genetic reinstatement approaches starting at various developmental stages in mouse models of epileptic syndromes caused by pathogenicity of Mecp2, Scn1a, Syngap1, Fmr1, and Ube3a (distinct curves for audiogenic seizures and pro-epileptogenic phenotype). Arrowheads denote different developmental stages at the time of rescue.
Another fascinating finding of this study is that even suboptimal Scn1a gene reinstatement was sufficient to eliminate the occurrence of spontaneous recurrent seizures and related mortality. Quantitative Scn1a expression analysis suggested two Scn1a Stop/+ -Cre mice had limited (<50%) protein rescue in cortices or hippocampi. However, this suboptimal or minimal Scn1a re-expression was sufficient to completely halt hyperthermic seizures and the development of spontaneous seizures. This observation is also consistent with the fact that injection of AAV-PHP.eB-Cre into Scn1a Stop/+ mice at P90 can eliminate the spontaneous recurrent seizures starting from ∼1 week after the virus delivery, which is much shorter compared to the typical time frame (∼3–4 weeks) required for optimal transduction of AAV-PHP.eB and cargo expression. These unexpected results suggest that an epileptic rescue threshold, which is below 100% SCN1A restoration, may exist. Thereby, in addition to this well-designed temporal rescue study, a systematic dosage rescue study is essential to leverage the amount of SCN1A gene product that is required to achieve complete seizure control. Knowing both the temporal and dosage therapeutic ranges is critical for guiding an effective and safe clinical trial.
In contrast to the full rescue of seizure and SUDEP phenotypes, the majority of behavioral abnormalities tested in this study were only partially rescued in terms of significance and/or magnitude by Scn1a reinstatement beyond symptom onset. The different efficacy between seizure and behavioral rescue suggests a critical role of Scn1a during early development in regulating the motor, social and cognitive functions in adulthood. Another possible explanation is that the early exposure to repeated seizures contributes to or exacerbates the behavioral abnormalities that persist into adulthood.
This proof-of-concept study extends the temporal “therapeutic window” of DS and informs the design of ongoing and future clinical trials leveraging SCN1A restoration. Although it is challenging to equate postnatal stages of mouse and human development, the authors push the therapeutic window into at least P90 in mice, translating to the possible full spectrum of seizure recuse ranging from infancy to adulthood in human DS individuals. This finding is extremely critical because it demonstrates that full seizure rescue is possible via gene therapy well beyond the time of epileptic symptom onset in DS.