
Abstract
Harper CB, Small C, Davenport EC, Davenport EC, Low DW, Smillie KJ, Martínez-Mármol R, Meunier FA, Cousin MA. J Neurosci. 2020;40(23):4586-4595. doi: 10.1523/JNEUROSCI.0210-20.2020 The epilepsy-linked gene SV2A has a number of potential roles in the synaptic vesicle (SV) life cycle. However, how loss of SV2A function translates into presynaptic dysfunction and ultimately seizure activity is still undetermined. In this study, we examined whether the first SV2A mutation identified in human disease (R383Q) could provide information, regarding which SV2A-dependent events are critical in the translation to epilepsy. We utilized a molecular replacement strategy in which exogenous SV2A was expressed in mouse neuronal cultures of either sex, which had been depleted of endogenous SV2A to mimic the homozygous human condition. We found that the R383Q mutation resulted in a mislocalization of SV2A from SVs to the plasma membrane but had no effect on its activity-dependent trafficking. This SV2A mutant displayed reduced mobility when stranded on the plasma membrane and reduced binding to its interaction partner synaptotagmin-1 (Syt1). Furthermore, the R383Q mutant failed to rescue reduced expression and dysfunctional activity-dependent trafficking of Syt1 in the absence of endogenous SV2A. This suggests that the inability to control Syt1 expression and trafficking at the presynapse may be key in the transition from loss of SV2A function to seizure activity.
Commentary
The very end of a neuron’s axon is jam-packed with fancy machinery designed to launch neurotransmitter into the synapse at a moment’s notice. Remarkably, in healthy individuals, this neurotransmitter release machinery operates endlessly to ensure that the last millisecond in the life of a neurotransmitter-containing synaptic vesicle—to borrow from the title of an excellent review on the subject 1 —is a successful one. Understandably, however, if a single component of the complex release machinery is flawed, then devastating consequences often result.
One component of the release machinery that has garnered much attention in epilepsy research is Synaptic Vesicle Protein 2 (SV2). Multiple lines of evidence demonstrate that SV2 plays an important role in defining the excitability of neural circuits that produce seizures. First, a single point mutation (R383Q) in SV2A, the only SV2 paralog expressed by inhibitory neurons, has been identified in a patient with intractable epilepsy. 2 Second, deletion of the Sv2a gene in mice results in severe and lethal seizures. 3 Finally, levetiracetam (aka Keppra), a highly effective anti-seizure drug, binds to and modulates SV2A to treat epilepsy. 4 With such evidence, one might assume that resolving how SV2A dysfunction contributes to epilepsy would be straightforward. Reality, however, has not borne out this assumption.
So, what is SV2A? The protein gets its name from its close association with synaptic vesicles, neurotransmitter-containing, secretory organelles found at presynaptic axon terminals (Figure 1A). 5 Prior to release, many vesicles sit docked at the axon terminal, a process involving the association of protein complexes called SNAREs that are found on the membranes of both vesicles (ie, vesicular-SNAREs) and the axon terminal (ie, target-SNAREs). 1 Remarkably, the close association of v- and t-SNAREs results in the molecular equivalent of closing a zipper that brings vesicle and axon membranes in close apposition (Figure 1A). These zippered-up vesicles are now primed and await fusion with the membrane of the axon terminal, an event that produces a pore opening that enables neurotransmitter molecules to pour into the synapse. Vesicle fusion—quite literally, the fusion of the primed vesicle’s membrane with the axon terminal’s membrane—is triggered by the influx of calcium through voltage-gated calcium channels that open in response to the strong depolarization provided by an incoming action potential (Figure 1A). Synaptotagmin, a protein found on the membrane of the vesicle, serves as the calcium sensor that enables vesicle fusion during calcium-dependent neurotransmitter release. 1 Resolving these complex processes, only superficially described here, ultimately resulted in the 2013 Nobel Prize in Physiology or Medicine shared by Thomas Südhof, James Rothman and Randy Schekman.
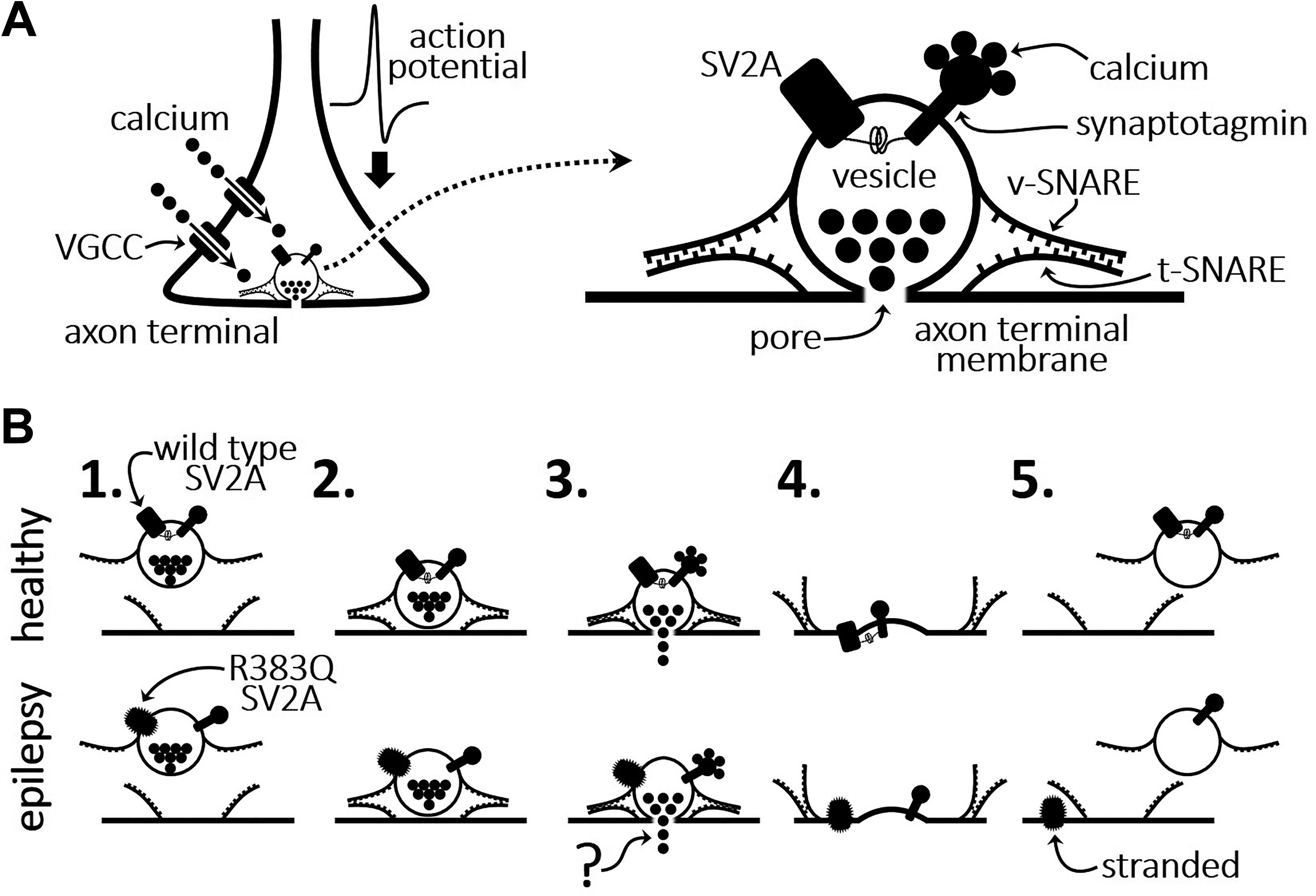
The R383Q mutation alters the trafficking of synaptic vesicle 2A (SV2A) and synaptotagmin. A, Schematic of an axon terminal (left). The strong depolarization of an action potential invading the axon terminal opens voltage-gated calcium channels (VGCCs). The influx of calcium triggers the release of neurotransmitter from vesicles docked at the membrane of the axon terminal (right). SNARE protein complexes on the membranes of the vesicle (v-SNARE) and axon terminal (t-SNARE) associate in a zipper-like manner that brings the vesicle in close apposition to the terminal membrane. When calcium binds to synaptotagmin, an integral vesicular protein that functions as a calcium sensor, a pore forms in the docked vesicle and neurotransmitter is released. SV2A is proposed to function as an intrinsic trafficking partner (iTRAP) for synaptotagmin. 5 B, The steps of neurotransmitter release in healthy (top row) and in the epilepsy-associated, R383Q SV2A mutation (bottom row). (1) A neurotransmitter-containing vesicle is trafficked to the axon terminal membrane. (2) v- and t-SNAREs associate and draw the vesicle closer to the terminal membrane. (3) Calcium binds to synaptotagmin to cause pore formation. (4) The membrane of the vesicle integrates with the terminal membrane. (5) A new vesicle is generated by endocytosis. In healthy individuals, SV2A carries synaptotagmin back to the newly formed vesicle. This process is dysfunctional in the R383Q mutation. How the mutation affects neurotransmission remains unclear.
SV2A, then, is one of many integral proteins found on the membrane of synaptic vesicles that enables successful neurotransmitter release (Figure 1A). That much is clear from SV2A knockout mice, wherein the release of vesicles containing the inhibitory neurotransmitter, γ-aminobutyric acid (GABA), from axon terminals in the hippocampus is significantly reduced. 3 Synaptic vesicle 2A deletion in mice also reduces the number of releasable, catecholamine containing vesicles from chromaffin cells, 6 endocrine cells that rely on release machinery similar to that used by neurons. But what role does SV2A play in neurotransmitter release, and how does SV2A dysfunction result in severe epilepsy? While the answers to these questions remain unclear, one common thread of years of work on SV2A points to an interaction with synaptotagmin, 7,8 the aforementioned calcium sensor. The authors of a recent study now provide new clues.
By focusing on the R383Q mutation in SV2A, Harper et al 9 show that the epilepsy-associated point mutation causes the protein to spend too much time on the membrane of the axon terminal and not on its rightful spot on the membrane of the vesicle; SV2A is essentially left stranded on the terminal membrane (Figure 1B). To understand how this might arise, one must consider that one of SV2A’s many proposed roles is that of an intrinsic trafficking partner (iTRAP). 5 After a synaptic vesicle fuses with the axon terminal membrane and releases neurotransmitter, the membrane of the vesicle and its associated integral proteins are retrieved and placed into a newly forming vesicle capable of repeating the whole process again. Intrinsic trafficking partners serve to retrieve integral vesicular proteins, and SV2A is proposed to function as an iTRAP for synaptotagmin. 5,8 By using a genetically encoded fluorescent pH reporter fused to SV2A, the authors show that the R383Q mutation appears to interfere with SV2A’s ability to traverse back to the reforming vesicle, and therefore, the mutation compromises SV2A’s ability to efficiently function as an iTRAP (Figure 1B). R383Q SV2A, the authors go on to show, is generally less mobile.
As expected, in the complete absence of SV2A, synaptotagmin accumulates on the axon terminal membrane, an observation recapitulated when the binding between intact SV2A and synaptotagmin is compromised. 7 Counterintuitively, however, the abnormal accumulation of synaptotagmin on the terminal membrane is also associated with the acceleration of synaptotagmin retrieval back to the vesicle, an observation that may reflect the recruitment of a synaptotagmin retrieval mechanism with kinetics faster than SV2A. 7 The R383Q SV2A mutant shows a similar phenotype: reduced binding with synaptotagmin and accelerated retrieval of vesicular synaptotagmin. 9 Additionally, the authors demonstrate that the R383Q mutation reduces the overall expression levels of synaptotagmin. Thus, the epilepsy-associated R383Q SV2A mutation both reduces synaptotagmin expression and causes any remaining synaptotagmin to be abnormally localized.
How the R383Q mutation ultimately causes epilepsy remains unknown. Considering that SV2A is the primary paralog in GABAergic neurons, one might be quick to conclude that the mutation reduces inhibitory neurotransmission, thus altering the overall balance between synaptic excitation and inhibition in favor of seizure-promoting excitation. Indeed, introducing a missense mutation in the Sv2a gene (L174Q) can reduce the release of GABA 10 ; notably, this conclusion was derived from in vivo microdialysis experiments, not from direct measurements of synaptic inhibition. However, a few findings complicate the conclusion that seizures associated with SV2A mutations result solely from altered GABAergic transmission. First, unlike the Sv2a-null mouse, the L174Q SV2A rat does not exhibit spontaneous seizures. Indeed, the animals appear normal until provoked with pentylenetetrazol. 10 Relative to control animals, less pentylenetetrazol is required to provoke seizures in the L174Q SV2A rat, suggesting that neural circuits in the mutant animal are primed to produce seizures. Second, in hippocampal culture systems, SV2A deletion enhances synaptic excitation while leaving synaptic inhibition intact 11 ; SV2A is also expressed by excitatory neurons. While these results are not generally incompatible with attempts to relate the epilepsy-causing, R383Q SV2A mutation to compromised synaptic inhibition, they do indicate that the story will likely be more complicated. That said, it is worthwhile digging in as new epilepsy-causing mutations in SV2A are being discovered. 12