
Abstract
Tewari BP, Chaunsali L, Campbell SL, Patel DC, Goode AE, and Sontheimer H. Nat Commun. 2018;9(1):4724. doi:10.1038/s41467-018-07113-0 Patients with brain tumor commonly present with epileptic seizures. We show that tumor-associated seizures are the consequence of impaired GABAergic inhibition due to an overall loss of peritumoral fast-spiking interneurons (FSNs) concomitant with a significantly reduced firing rate of those that remain. The reduced firing is due to the degradation of perineuronal nets (PNNs) that surround FSNs. We show that PNNs decrease specific membrane capacitance of FSNs permitting them to fire action potentials at supraphysiological frequencies. Tumor-released proteolytic enzymes degrade PNNs, resulting in increased membrane capacitance, reduced firing, and hence decreased GABA release. These studies uncovered a hitherto unknown role of PNNs as an electrostatic insulator that reduces specific membrane capacitance, functionally akin to myelin sheaths around axons, thereby permitting FSNs to exceed physiological firing rates. Disruption of PNNs may similarly account for excitation-inhibition imbalances in other forms of epilepsy and PNN protection through proteolytic inhibition may provide therapeutic benefits.
Commentary
The currently highlighted manuscript represents the latest study from a group who has extensively contributed to the knowledge in the field regarding the mechanisms underlying tumor-associated epilepsy, expanding our knowledge regarding the underlying pathological mechanisms (Figure 1). They initially discovered that glioma cells release excitotoxic concentrations of glutamate which acts in an autocrine fashion to increase the proliferation of glioma cells and induces excitotoxicity in the peritumoral region, thereby performing dual roles to facilitate and accommodate tumor growth, respectively (for review see reference de Groot et al 1 ). They have pinpointed the source of glutamate release from glioma cells to the system Xc− transporter (SXC; for review see reference de Groot et al 1 ). This group has also demonstrated that GABAergic signaling in the peritumoral region is compromised resulting from a loss of parvalbumin-positive (PV+) interneurons in the peritumoral region, compromised function of remaining fast-spiking interneurons (FSNs), and a shift in the equilibrium potential for chloride in excitatory neurons, due to a decreased expression of the K+/Cl− cotransporter. 2 Collectively, this group has contributed a large body of work demonstrating a 2-hit model of excessive glutamate release and compromised GABAergic inhibition contributing to tumor-associated epilepsy (Figure 1).
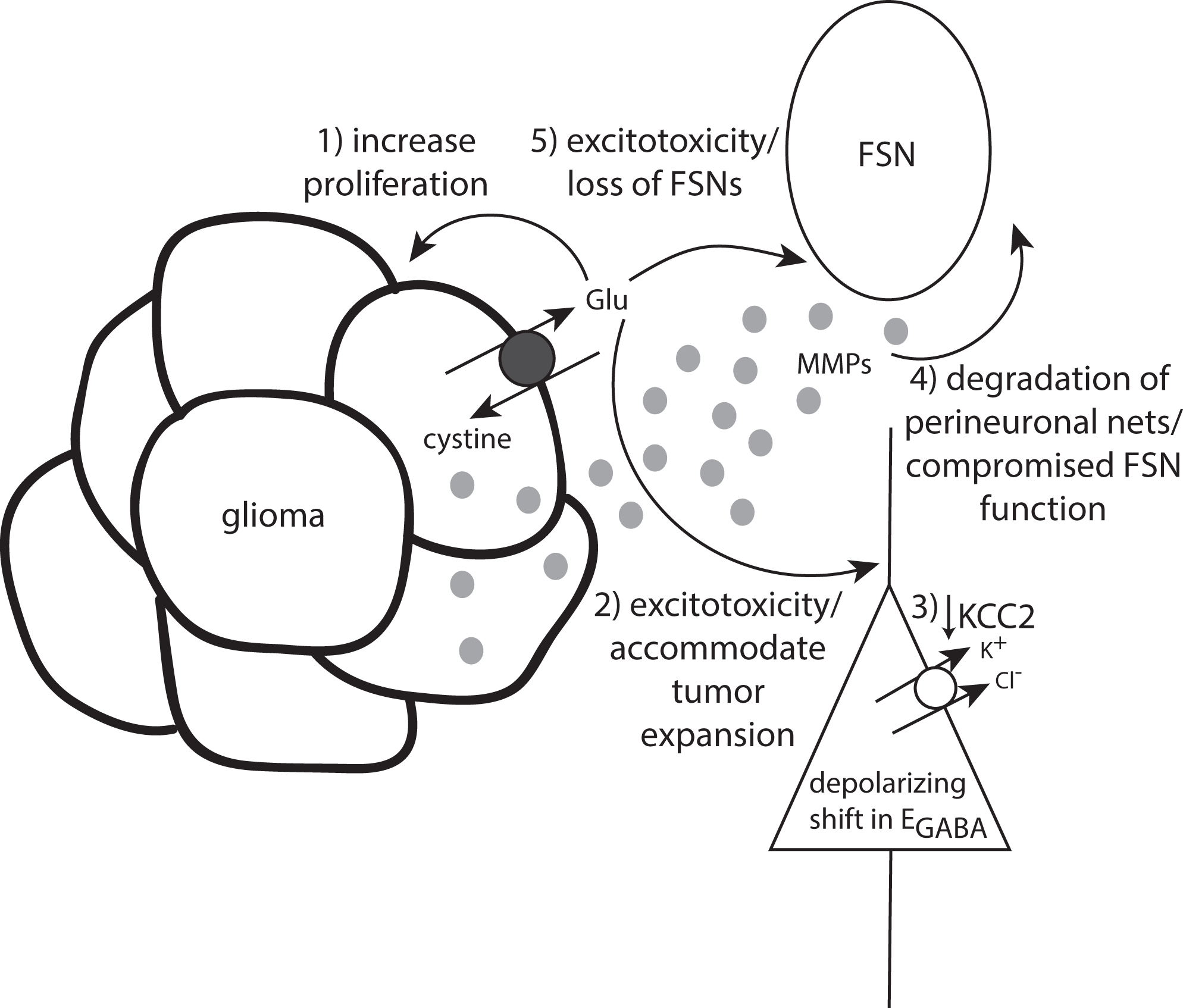
Numerous mechanisms whereby glutamate release from glioma cells influences tumor-associated epilepsy. Glutamate release via reversal of the system xc− transporter 1) increases the proliferation of glioma cells, 2) induces excitotoxicity in the peritumoral region facilitating tumor expansion, and 3) reduces KCC2 expression in principal neurons causing compromised GABAergic inhibition. Glioma cells also release MMPs leading to 4) degradation of perineuronal nets and 5) loss of fast-spiking interneurons.
In the currently highlighted manuscript, the authors demonstrate that glioma cells release matrix metalloproteinases (MMPs), degrading perineuronal nets (PNNs) surrounding FSNs in the peritumoral region, compromising GABAergic inhibition and contributing to tumor-associated epileptiform activity. These findings propose a mechanism underlying the observed loss in FSNs and the compromised function of remaining FSNs in the peritumoral region. In addition, previous studies point to a role for extracellular matrix proteins in establishing the chloride gradient required for effective inhibitory GABAergic signaling, 3 which suggests that the degradation of PNNs may also underlie the observed changes in chloride homeostasis in the peritumoral region.
A loss of FSNs has been demonstrated previously in numerous models of brain trauma, including tumor models, ischemia, and Traumatic brain injury (TBI). In fact, a degradation of PNNs surrounding PV+ interneurons has also been demonstrated following TBI. 4 Perineuronal nets are thought to protect FSNs from oxidative stress and excitotoxicity, 5 and thus, the degradation of PNNs increases vulnerability to the loss of FSNs. Despite similarities in the neuropathology in these models, the authors of the current study suggest that release of MMPs from glioma cells, not astrocytes, mediate the loss of FSNs in the peritumoral region, which obviously does not translate to the other models of brain injury. This evidence would suggest that despite the similar neuropathology, there are unique mechanisms contributing to the loss of FSNs in these models of brain injury. To our knowledge, the mechanisms underlying the loss of PNNs in other models of brain injury, such as TBI, remains unknown.
An alternative interpretation of these results is that the degradation of PNNs surrounding PV+ interneurons is a beneficial mechanism to support the increased plasticity necessary for recovery, as has been proposed in the case of ischemia. 6 This theory builds on the well-established role for PNNs in neural plasticity (for review see reference Wang et al 7 ). Perineuronal nets play a role in regulating plasticity during development and into adulthood. The opening of PNNs is thought to facilitate synaptic plasticity during critical periods; whereas, the establishment of PNNs is thought to stabilize existing connections and limit further plasticity. Thus, it is possible that the degradation of PNNs in the peritumoral region may play a role in homeostatic plasticity to help recovery and the restoration of appropriate synaptic transmission following injury. Although this is an attractive theory, data presented in the currently highlighted manuscript directly demonstrates that degradation of PNNs with chondroitinase ABC (ChABC) increases epileptiform activity. Further, ChABC has been shown to increase seizure susceptibility. 8 In addition to the role of PNNs in plasticity, they are also thought to confer protection to oxidative stress 5 and, therefore, may also play a role in neuroprotection. As such, disruption of PNNs has been observed under pathological conditions and has been implicated in numerous disorders, ranging from Alzheimer disease to addiction (for review see reference Sorg et al 9 ). The currently highlighted manuscript suggests a role for PNNs in the underlying neuropathology of tumor-associated epilepsy, in which degradation of PNNs surrounding FSNs leaves these neurons vulnerable to neurodegeneration and compromised function in those that remain. Combined, these findings demonstrate that the degradation of PNNs contributes to hyperexcitability rather than supporting recovery of the network following injury.
Consistent with the notion that the degradation of PNNs contributes to hyperexcitability, the authors demonstrate compromised function of remaining FSNs associated with changes in the electrophysiological properties of these neurons, including the inability of sustain their characteristic high firing rate. Fast-spiking interneurons in the peritumoral cortex exhibit a higher membrane capacitance, a change which the authors pinpoint to the loss of the surrounding PNN, since it can be mimicked with ChABC and blocked using an MMP inhibitor. The authors suggest that PNNs subserve a previously unknown function, acting in similar fashion to oligodendrocytes forming a myelin sheath to decrease the membrane capacitance of FSNs, thereby facilitating the high firing rate of these neurons. This is a provocative idea and supports a novel function for PNNs. However, since this finding is not the primary focus of the current study, it is not given the attention that it deserves. Although it may be a bit premature to ascribe this essential function to PNNs, the evidence presented in the currently highlighted manuscript supports this notion and is certainly worthy of further study.
The currently highlighted manuscript is the latest study contributing to an extensive body of work from the Sontheimer laboratory characterizing mechanistic changes contributing to tumor-associated epilepsy, summarized in Figure 1. This line of research has led to the identification of the SXC, which contributes to glutamate release from glioma cells contributing to tumor expansion and tumor-associated epilepsy (for review see reference Sontheimer et al 10 . Sulfasalazine, which blocks SXC, has shown to be effective in preclinical glioma models and is being pursued in clinical trials for the treatment of glioblastoma. In addition, the currently highlighted study also points to defective inhibitory signaling in tumor-associated epilepsy due to the degradation of PNNs and suggests that MMPs may be a useful therapeutic target.