
Abstract
Introduction:
Gene therapy encompasses a diverse array of genetically engineered products in biomedical research. As novel products continue to gain regulatory approval, institutions will be challenged by translating research processes into the clinical environment. This article will provide a summary of the 5 in vivo viral-based therapies that have been approved or are under review in the United States or European Union and discuss the development of biosafety handling practices in the clinical setting.
Discussion:
Commercially approved gene therapies utilize adeno-associated viral vectors, lentiviruses, and modified herpes simplex viruses for genetic manipulation. Health care personnel must understand the location of the genetic manipulation, ex vivo or in vivo, in order to develop safe work practices when handling the products. Occupational exposure to a viral agent could lead to risks of infection or acquired immunity. Institutions must merge biosafety and hazardous drug handling standards in order to develop safe handling procedures for clinical care.
Conclusion:
As biotechnology continues to advance, so will the challenges of incorporating novel therapies into the clinical setting. Health systems must educate themselves on the current recommendations and maintain competency of this evolving science to ensure the safety of patients, families, and staff in the clinical environment.
Introduction
Gene therapy is on track to revolutionize the landscape of modern medicine. These novel drugs are bringing hope to the rare disease community while simultaneously promising to rewrite the natural history of those affected. Spinal muscular atrophy patients as well as those with rare retinal dystrophies have witnessed life-changing effects, ranging from improved quality of life to an extension in their overall life expectancy, all with a single infusion. 1,2 These 2 patient populations represent a small subset of many hopeful gene therapy candidates. Over 700 new gene therapy trials have been initiated worldwide in just the past 5 years, which may signify a potential surge of new drug approvals by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) in the near future. 3
In the broadest sense, gene therapy encompasses a diverse array of genetically engineered products in biomedical research. 4 By targeting diseases at the genetic level, gene therapy provides clinicians the ability to treat the condition itself rather than solely mitigating the disease symptoms through traditional medications. Tisagenlecleucel, an autologous T-cell immunotherapy, was lauded as the first gene therapy drug approval in the United States back in 2017. 5 This cellular-derived therapy utilizes a modified lentivirus as a vector to reprogram autologous T-cells. The T-cells are programmed to directly target the patient’s leukemia cells. 6 This genetic manipulation occurs ex vivo as the cells are harvested from the patient and then genetically engineered at a manufacturing facility. 6,7 Cells are thawed, washed, transduced with the CD19 transgene at the facility, expanded, and then cryopreserved before the final gene therapy product is returned to the clinical site for patient administration. 6,7
Although these cellular-derived gene therapies involve genetic modification, their risk profile may differ from commercial therapies that alter genetic makeup in vivo. Health care personnel must understand the location of the genetic manipulation, ex vivo or in vivo, in order to develop safe work practices when handling the products. Commercially approved cellular gene therapies, such as tisagenlecleucel and axicabtagene ciloleucel, arrive at the clinical site as a ready-to-infuse formulation. 6,8 After the ex vivo genetic manipulation at a manufacturing facility, these 2 cellular therapies require certification to be free of any viral pathogens. 9 Without any known viral pathogens associated with the product, handling procedures for these autologous cellular therapies could mirror procedures for human specimen handling or processing at the clinical level. The bloodborne pathogen standards from the Occupational Safety and Health Administration (OSHA) provide safeguards to prevent the occupational exposure of blood or other potentially infectious materials in the workplace. 10
In contrast, commercial viral-mediated gene therapies, such as onasemnogene abeparvovec and voretigene neparvovec, modify genetic material in vivo. 1,2 This is accomplished via a pharmaceutical injection of an adeno-associated viral vector. 1,2 These 2 viral-vector therapies, in addition to a genetically modified oncolytic viral therapy that will be discussed later in this review, require on-site clinical manipulation and therefore present a newly emerged biosafety risk in the health care environment. 1,2,11 -13 This article will provide a summary of the 5 in vivo viral-based gene therapies that have been approved or are under review in the United States or European Union and discuss the development of biosafety handling practices in the clinical setting.
Discussion
Commercial In Vivo Viral-Based Therapies
Talimogene laherparepvec
Talimogene laherparepvec, or T-Vec, is an oncolytic viral therapy (a therapy that targets cancer cells) that is composed of a live, attenuated herpes simplex virus (HSV) type 1. 11,14 This therapy is administered via a direct intratumoral injection into palpable or visible lesions of melanoma patients. 11,14 Through genetic modification, T-Vec is able to replicate within the tumor cells and produce proteins called granulocyte-macrophage colony-stimulating factors, or GM-CSF, which together, with the release of tumor-derived antigens, illicit an immune response. 14,15 In the Phase 3 pivotal trial, 64% of T-Vec injected lesions decreased in size, with 48% showing a complete response to treatment. 16 T-Vec was the first oncolytic immunotherapy approved globally, with the FDA granting permission in October 2015 and the EMA in December 2015. 14,17,18
Alipogene tiparvovec
Alipogene tiparvovec was the earliest of the viral-vector therapies on the market, gaining EMA approval in October 2012 for patients with lipoprotein lipase deficiency (LPLD)—an autosomal recessive disease that causes an inability to break down fat. 19 -21 Patients suffering from LPLD are at a high risk of severe, sometimes life-threatening pancreatitis. 21 Alipogene tiparvovec utilized an adeno-associated virus serotype 1 vector to introduce a functional copy of the LPLS447X gene into affected muscle cells. 20 Patients in clinical trials saw a 40% or more decrease in fasting triglycerides between weeks 3 and 12 postinjection. 22 Unfortunately, the rarity of the disease eventually led the manufacturer to allow their EMA authorization to lapse in October 2017 and discontinued further marketing of this treatment. 22,23
Voretigene neparvovec
The same month of alipogene tiparvovec’s authorization lapse, a key FDA advisory panel made history by unanimously recommending approval of the first viral vector treatment in the United States. 24 Two months later, the FDA agreed and approved commercialization of voretigene neparvovec. 25 Developed by Spark Therapeutics, this live, nonreplicating adeno-associated viral vector serotype 2 delivers the human retinal pigment epithelial protein into the subretinal space of affected patients. 1 Efficacy of this gene therapy was established through the use of multiluminance mobility testing (MLMT). 1,26,27 This test measures an individual’s ability to reasonably and accurately navigate a course at different levels of illumination; an MLMT score of 2 or greater represents clinically meaningful benefit. 26 More than 50% of the subjects enrolled in the active gene therapy arm had an MLMT change of 2 or greater at the 1 year follow-up, whereas only 10% of the control group had an MLMT change of at least 2. 1,27 Voretigene neparvovec remains on the market as a viable treatment option for patients with this rare retinal dystrophy.
Onasemnogene abeparvovec
Onasemnogene abeparvovec is an adeno-associated virus serotype 9 vector that has been genetically modified to carry the human survival motor neuron 1 (SMN1) gene. 2 Marketed under the trade name Zolgensma, this gene therapy medication was approved by the FDA in 2019 for children with spinal muscular atrophy (SMA) up to the age of 2 years. 2 Children with SMA type 1 typically fail to meet motor milestones and will either progress to mechanical ventilation or death by the age of 2 years. 28 Initial studies have found that a single dose of onasemnogene abeparvovec resulted in a rapid improvement of motor functions over baseline compared to a natural history cohort—particularly for those patients treated earlier in life. 29,30 At 24 months postinfusion, all 12 patients treated under the high dose cohort in the initial clinical trial remained alive and free of permanent ventilation. 2,29 Additional clinical trials are ongoing or planned that will continue to explore the use of onasemnogene abeparvovec in older populations.
Valoctocogene roxaparvovec
Although the earliest viral-vector mediated gene therapies all targeted rare diseases, valoctogene roxaparvovec is an emerging treatment that could change that course. This novel therapy for Hemophilia A—a blood disease with an incidence of approximately 1 in 4000 to 5000 live births—utilizes an AAV serotype 5 vector to introduce the B-domain deleted human factor VIII transgene into affected patients. 31,32 Current available clinical trial data show a 96% reduction in an individual’s mean annualized bleeding rate, a favorable safety profile, and good tolerance by patients across all doses. 31,32 Valoctocogene roxaparvovec has been granted both Breakthrough Designation and Orphan Drug Designation by the FDA and is currently under review for approval. 33
Development of Institutional Handling Procedures
Viral-based in vivo therapies represent an emerging class of pharmaceuticals. However, with only 3 formulations on the commercial market, their limited use and novel nature suggest an education opportunity for health care workers. Although the Centers for Disease Control and Prevention and the National Institute of Health have published guidance to assess risk and conduct of safe work environments with viral agents and recombinant nucleic acid products in the laboratory and research settings, no similar guidelines or formal processes exist for viral-based therapies in clinical care. 12,34 -36
Basic handling information for a viral-based therapy can be found in the prescription drug labeling, also referred to as a package insert or medication guide, of each pharmaceutical product. In the US, this information is approved at the time of marketing and provides a comprehensive drug summary for the pharmacist or clinician and includes information on dosing, administration, clinical pharmacology, approved indications, and toxicities. Title 21, Part 201 of the Code of Federal Regulations provides the specific content requirements for the labeling of any prescription drug and biological product. 37
Health systems may wish to standardize biosafety practices across the continuum of viral-mediated gene therapies for ease of policy development, but this practice may prove difficult due to inconsistencies found in the published drug labeling. Only 2 of the 3 commercially available in vivo gene therapies contain references to biosafety risks in their package inserts (Table 1). 1,2,12 Therefore, biosafety guidance for viral-mediated pharmaceutical products should be drawn from established laboratory standards in Biosafety in Microbiological and Biomedical Laboratories (BMBL) and NIH Guidelines for Recombinant DNA Research. 12,35,36 The biosafety standards used for the pharmaceutical formulation should, at minimum, align and meet the biosafety level containment practices for the associated viral agent. 12
Commercially Available Gene Therapies in the United States Utilizing In Vivo Genetic Manipulation (as of 6/2020).

In addition to the containment practices for biosafety, institutional policy must also consider the clinical standards for pharmaceutical compounding and hazardous drug handling. The United States Pharmacopeia (USP) is a nonprofit scientific organization that sets federally recognized quality and safety standards (Table 2) for nonsterile and sterile compounding, the handling of hazardous drugs, and radiopharmaceuticals. 38,39 Although USP is not considered an enforceable body, its standards are often enforced by various other groups, including the Joint Commission, states and their respective boards of pharmacy, and the FDA. 38 -40 It is critical for health care settings to maintain compliance with the parameters set forth by this group.
United States Pharmacopeia Healthcare Quality & Safety Standards (as of 6/2020).

USP General Chapter 800 (USP<800>), Hazardous Drugs—Handling in the Healthcare Setting, was developed in conjunction with USP<795> and USP<797> standards to ensure the occupational safe handling of hazardous drugs (HD). 41 USP<800> requires that institutions maintain an HD list. This list must include all drugs deemed hazardous by the National Institute of Occupational Safety and Health (NIOSH) and should consider investigational drugs and newly approved drugs with limited occupational safety data. 41 -44 Although the NIOSH criteria can be used to identify drugs with mutagenic, carcinogenic, or teratogenic properties, it falls short of identifying biologic drugs as hazardous. 39,42 Current NIOSH standards only assess the hazardous potential of drugs approved by the Center for Drug Evaluation and Research (CDER) and do not evaluate hazardous occupational risks of handling gene therapies, which are approved by the Center for Biologics Evaluation and Research (CBER). 45
Both USP<800> and NIOSH recommend that institutions assess all drugs utilized within their health system for occupational hazards. As such, viral-based in vivo therapies should be added to an institution’s hazardous drug list due to the established and theoretical occupational risks for handling these therapies. 12,46 For example, the package insert of T-Vec indicates that health care personnel that are immunocompromised or pregnant should not handle, prepare, or administer the medication due to risk of developing a herpetic infection. 11 Onasemnogene abeparvovec recommends that caregivers practice safe handling of bodily fluids due to the risk of viral shedding. 2 Contact precautions for onasemnogene abeparvovec should continue for 30 days postinfusion to avoid any theoretical transmission of the virus. 2 Transmission could lead to unintended antibody development that would prevent a sibling or close contact of a treated patient from being eligible to receive their own gene therapy infusion due to this acquired immunity. 12
In addition, the mutable nature of viruses should not be discounted. Although highly unlikely, there is the potential for the recombination of vector virus with wild-type parental viruses within the treated patient’s cells. This recombination may result in an antigenic shift, changing the virus’s characteristics. Such a phenomenon is believed to be linked to the H1N1 influenza virus in 1918, which led to the Spanish Flu pandemic outbreak. 47 Other potential risks include the reactivation of a “disabled” virus’s characteristics, such as replication. Replication increases the risk of transmission and the possible creation of transgenes or viral oncogenes. 48 Furthermore, any introduction of foreign genetic material during drug production has the potential to change the final viral outcome. 48 Quality assurance testing is necessary to protect the authenticity of the product.
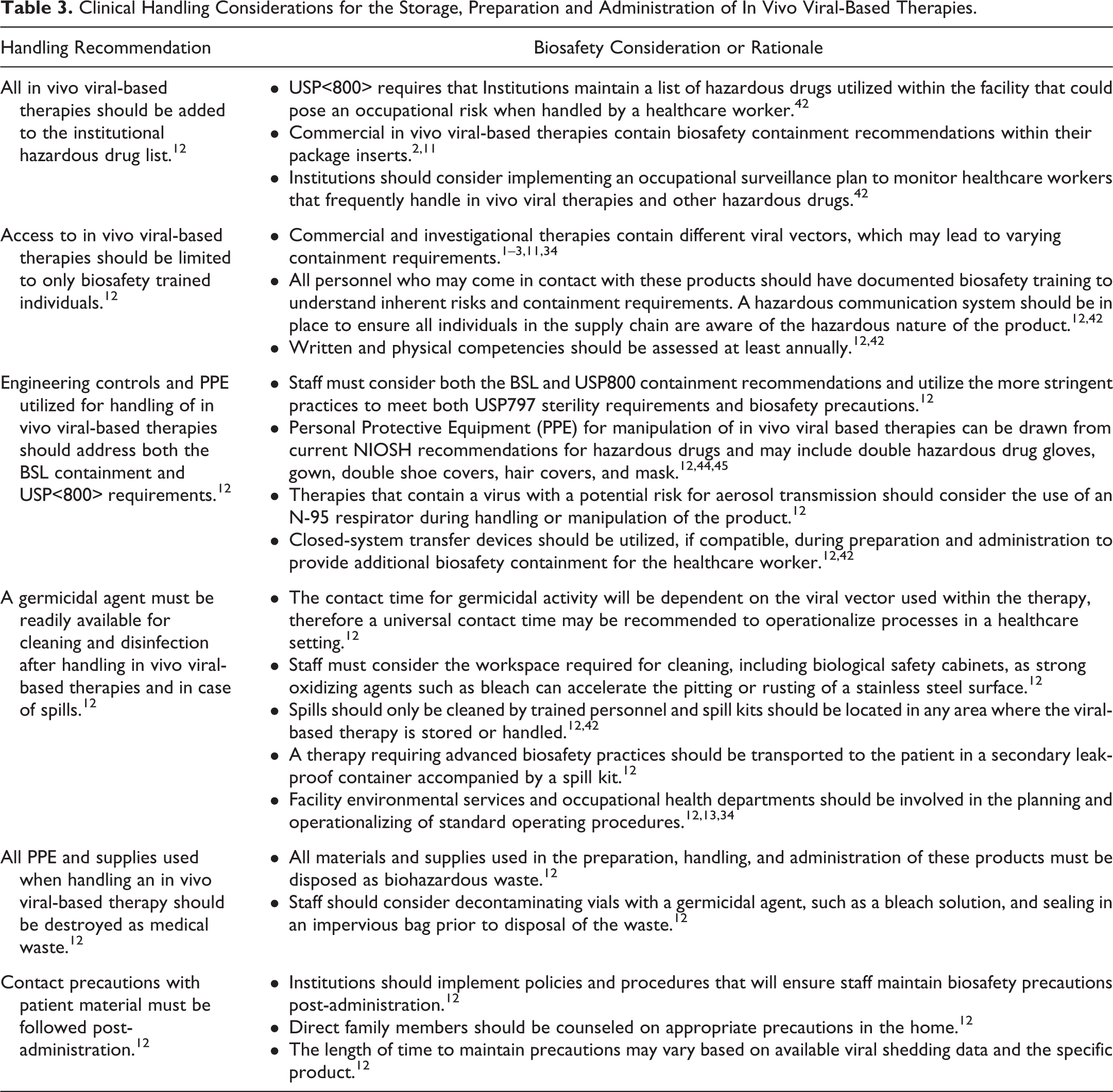
USP<800>, NIOSH, and OSHA all provide guidance on handling hazardous drugs in the workplace. These clinical guidance documents set the framework for policy development and personal protective equipment when manipulating or administering biohazardous drugs. It is important to note that the recommendations within these clinical documents often exceed the minimum biosafety level containment strategies within the BMBL, NIH guidelines, and OSHA bloodborne pathogen standards. However, it may be necessary for an institution to comply with the more stringent recommendations in order to be compliant in the clinical setting. Health systems must plan to rely on biosafety, epidemiology, and pharmaceutical experts within the institution to fully assess the occupational risks of handling a pharmaceutical product that contains a viral vector or recombinant nucleic material. 12,13 Table 3 provides a summary of handling recommendations for in vivo viral-based therapies in the clinical environment.
Clinical Handling Considerations for the Storage, Preparation and Administration of In Vivo Viral-Based Therapies.
Conclusion
As biotechnology continues to advance, so will the challenges of incorporating novel therapies into the clinical setting. Commercial drug labeling may contain varying levels of biosafety containment despite using a similar viral vector between 2 formulations. Therefore, health systems must educate themselves on the current guidance within both the clinical and biosafety community in order to ensure ongoing competency and safety in this evolving science. Key players should be identified at the institution to create and operationalize safe handling guidelines, including biosafety experts, epidemiologists, physicians, pharmacists, and occupational health staff. 12,13 These experts must commit to ensuring internal policy stays compliant and up to date with the latest guidance in their individual fields. This team approach to policy development and review will ensure the safety of patients, families, and staff in the clinical environment.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Jill E. Blind and E. Nicole McLeod hold licensing agreements with Sarepta Therapeutics, Prevail Therapeutics, Apic Bio, and bioStrategies Group related to the handling of viral-mediated therapies in a pharmaceutical setting.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.