
Abstract
Introduction:
Inactivation of biological agents and particularly select agents has come under increased scrutiny since the US Army inadvertently shipped live anthrax both inside and outside the US, leading to more stringent regulations regarding inactivation.
Methods:
Formalin and Trizol® LS were used to inactivate virus samples in complex matrices. Cytotoxic chemicals were removed using either desalting or concentrating columns or through dilution using HYPERFlasks. Efficacy of inactivation was evaluated either through plaque assay or immunofluorescence assay.
Results:
All virus samples and tissue specimens were successfully inactivated using either formalin or Trizol® LS. Both the desalting columns and concentrating columns were able to remove cytotoxic chemicals to facilitate viral amplification in controls. Dilution of cytotoxic chemicals through HYPERFlasks was also successful provided that media was changed completely within 48 hours of first cell passage.
Discussion:
All inactivation testing demonstrates that both formalin and Trizol® LS successfully inactivate virus-infected cell lines and tissues, which is consistent with previously published literature. Each sample cleanup method has its benefits and pitfalls. Desalting columns can process the largest sample size but are also susceptible to plugging and degradation, whereas concentrating columns are not as vulnerable but can only process 5% of the sample load per run.
Conclusion:
Based on our results along with those of our colleagues, it is recommended that the regulatory authorities re-evaluate the requirements for each entity to validate well-established inactivation methods in house because there would be limited benefits despite the considerable resources required for this effort.
Introduction
The safety considerations for manipulating biological agents either within or outside biocontainment laboratories following inactivation are of the highest importance. Biocontainment laboratory operations can be limited by space and regulatory burden, leading researchers to prefer to inactivate samples for further analysis outside of the biocontainment laboratory. 1 Inactivation of biological agents and particularly select agents has come under increased scrutiny since the US Army inadvertently shipped live anthrax to over 190 laboratories both inside and outside the US over many years up until 2015, leading to more stringent regulations regarding inactivation. 2
The latest version of the Select Agent Regulations requires an entity to inactivate any samples containing a select agent with a validated method. 3 Validation includes not only the method used to inactivate the agent but also viability testing of the sample to verify it is indeed killed. Although inactivation can be accomplished on many sample types with limited additional resources, viability testing following inactivation using Trizol® LS or formalin-based solutions requires resources and time because of the potential toxic effect of disinfectant residues on cell cultures required for virus infectivity testing. Trizol® LS solution is used for inactivation of various liquid samples through the denaturing of macromolecules taken from potentially infected individuals as well as stock virus. 4 Formalin-based solutions are effective at inactivating both viral and bacterial-based agents through the mechanism of generating DNA or protein-based cross-links. Samples can include plasma, urine, and more complex matrices such as semen or breast milk if independently validated. Formalin is used for inactivation of infected cell monolayers and tissues taken for pathogenesis studies. Inactivation of viruses using both formalin and Trizol LS-based solutions have been used for years safely.4 -9 Use of Trizol-based solutions was recommended by WHO for processing clinical samples from Ebola virus (EBOV) outbreak regions during the West Africa Outbreak in 2014.4 -7
Because validation of both inactivation methods requires significant resources, we sought to produce a series of validation studies in compliance with the latest Select Agent Regulations. 2 This was accomplished by evaluating new sample cleanup procedures and describing the amount of resources necessary to accomplish this process. This shows that for well-established methods, there are resource requirements including personnel labor hours, supplies, and containment laboratory space needs for research institutes to absorb with minimal additional benefits to both researchers and the public compared to what already exists in the literature. In this article, we will describe inactivation of virus-infected cell culture monolayers using formalin, formalin inactivation of tissue samples from virus-infected animals, and inactivation of virus stocks and biological fluids from animals infected with viruses using Trizol® LS solution. All viruses used in these studies are lipid enveloped viruses from different virus families requiring BSL-2, BSL-3, or BSL-4 containment for manipulations. Before use of columns/filters to remove caustic chemicals such as Trizol® LS from the sample prior to adding it to susceptible cells (viability testing), dilution of the sample has commonly been used. 4 The difficulty with dilution is that most chemicals such as Trizol® LS must be diluted at least 1:1000, increasing the number of flasks required for testing tenfold and also increasing the limit of detection. To handle this increased volume, we have explored the use of HYPERFlasks 10 and developed a strategy for testing Trizol LS-treated samples. HYPERFlasks require 560 mL of media/inoculum per flask and do not require an additional 1-hour incubation as typically used for T175 flasks or roller bottles. The advantage that HYPERFlasks present researchers is that it allows them to test and evaluate larger sample sizes compared with standard flasks, enhancing throughput for validation studies involving Trizol® LS or formalin-based solutions. Sample cleanup using either concentrating or desalting columns as well as the HYPERFlasks are also described.
Methods
Formalin Inactivation of Cell Monolayers
Vero E6 cells in 6 well plates were infected with MP12, a Risk Group 2 vaccine strain of Bunyaviridae Rift Valley Fever, at a multiplicity of infection (MOI) of 4.0 and incubated for 48 hours with 2 mL minimal essential media (MEM; Corning, catalog 10-010-CM) supplemented with 10% fetal bovine serum (FBS; GE Healthcare Life Sciences, catalog SH30071.03). Supernatant was then decanted, and 2 mL of 10% neutral buffered formalin (Fisher, catalog 245-684) was added per well. Plates were then placed at 4°C for 3.5 hours. The formalin was then decanted, and wells were washed 3 times with phosphate buffered saline (PBS) to remove residual formalin. To determine viability, cells were then scraped and collected in 300 uL of PBS and applied as an inoculum to Vero E6 cells in 6 well plates for 72 hours (P1) before passaging 66% of the supernatant from this set to another plate of Vero E6 cells for 72 hours (P2) to provide two passages for outgrowth of any potential virus. Presence of virus in the second passage was determined by using the supernatant from the final set of outgrowth plates as infection material in an immunofluorescence assay (IFA) as described by Mudhasani et al 11 (Figure 1).
Flow chart for virus inactivation of cell culture plates using formalin-based solutions. This flow chart demonstrates the infection, inactivation, and sample cleanup process for virus infected cell culture samples inactivated with formalin-based solutions. Fixation of plates with formalin solution was carried out at room temperature followed by storage at 4°C. All other incubations were carried out at 37°C as stated in the Methods section.
Two sets of positive controls were utilized. The first set of positive controls, 50 pfu per well, was applied to wells after receiving an inoculum of formalin-fixed cells to demonstrate the elimination of formalin and a hospitable environment for virus outgrowth should any viable virus have been present (Figure 2). The second set of controls used infected cells that were not subjected to formalin fixation and were also passaged to demonstrate viral propagation would continue under these conditions in the absence of formalin.

Flow chart for positive control cell culture samples. This flow chart demonstrates the infection and sample processing for virus-infected cell culture plates. All incubations were carried out at 37°C as stated in the Methods section.
Similar tests were carried out across multiple virus families to meet regulatory requirements of generating in-house data for each virus family to include Paramyxoviridae, Filoviridae, Arenaviridae, Togaviridae, and Coronaviridae with differing incubation periods for the outgrowth steps to accommodate differing viral replication rates.
Formalin Inactivation of Tissues and Removal of Formalin Using Concentrating Columns
Triplicate sets of livers from moribund guinea pigs succumbing to infection with Arenaviridae Lassa Josiah, a Risk Group 4 virus, were collected in 1-cm-thick sections and submerged in 10% neutral buffered formalin at ambient temperature for 20 and 29 days. Ratio of formalin solution to tissue was at least 10:1, with a tissue weight of approximately 1.0 g per sample. Unfixed tissues were also collected and stored at –80°C until the samples were processed. Tissues were collected in larger sizes for inactivation testing than would typically be harvested for pathology to represent a worst case scenario.
Tissues were removed from formalin and washed twice in 30 mL of PBS to remove excess formalin from the surface. The outer surface of the tissue was trimmed to retain the inner core to confirm formalin penetration and inactivation. Tissues were then homogenized and applied to an Amicon Ultra-15 Centrifugal Filter Unit cutoff columns (Stock Number UFC905008, Millipore Sigma, Burlington, MA) in 13 mL total volume of PBS. Samples were spun at 5000 × g until less than 1 mL of volume remained in the retained portion of the column. The columns were washed twice by applying 13 mL of PBS and spinning to again reduce volume to 1 mL or less, which functioned to remove both the cytotoxic chemicals from the formalin solution and the liver homogenate (Figure 3).
Flow chart for virus inactivation of animal tissues using formalin-based solutions. This flow chart demonstrates the infection, inactivation, and sample cleanup process for virus-infected animal tissues inactivated with formalin-based solutions. Fixation of tissues with formalin solution was carried out at room temperature followed by storage at 4°C. All other incubations were carried out at 37°C as stated in the Methods section.
To confirm the columns were capable of retaining virus, 2000 pfu of virus was applied to a set of columns containing uninfected tissue homogenate after the treatment described previously and underwent an additional 3 washes with PBS.
Washed homogenates were resuspended in PBS and applied as inoculum to Vero cells in 6 well plates for 3 days for an initial outgrowth stage (P1). Half of the supernatant was applied to a new set of Vero cells and incubated for an additional 3 days (P2). This second passage was assayed for viral content by IFA as described previously.
A duplicate set of uninfected livers was processed similarly without washing through a molecular weight cutoff column. Virus was spiked into test samples to evaluate whether virus would have grown in the presence of the test matrix without performing a cytotoxic cleanup (Figure 4). Similar experiments were completed with Filoviridae Zaire Ebolavirus.

Flow chart for processing of animal tissues as positive controls. This flow chart demonstrates the infection and sample processing for virus-infected animal tissues. All incubations were carried out at 37°C as stated in the Methods section.
Trizol® LS Inactivation of Stock Virus and Removal of Trizol® LS Using Concentrating Columns
Stock virus from the RG2 surrogate of Togaviridiae Venezuelan Equine Encephalitis, strain TC83, was concentrated to a titer of 4.4E+11 pfu/mL. Virus was combined with Trizol® LS (Thermo Fisher Scientific, catalog 10296028) at ratios of 3:1, 1:3, and 1:9 parts Trizol® LS to sample, or alternatively described as 75%, 25%, and 10% Trizol® LS final volume. Samples were then diluted 100 times in PBS for column compatibility (final Trizol® LS concentration 0.6% [v/v]) and applied to Amicon 100kD molecular weight cutoff columns and spun to concentrate to approximately 200 uL. Samples were then washed 3 times with 12 mL PBS and spun to concentrate each time.
To confirm the filter membranes were not compromised by the Trizol® LS, 500 pfu of virus was applied to a similarly treated column after the third wash and then washed 3 more times with PBS.
All contents of the final washed and concentrated samples were applied to Vero cells for 24 hours (P1). Greater than 50% of the supernatant was then passaged to a new set of Vero cells and allowed to propagate again for an additional 24 hours (P2). Samples from the second passage were then assayed by IFA for detection of viable virus (Figure 5).
Flow chart for Trizol® LS inactivation of stock virus and removal of Trizol® LS using concentrating columns. This flow chart demonstrates the inactivation and sample cleanup process for virus stocks inactivated with Trizol LS-based solutions utilizing the sample cleanup process with concentrating columns. All incubations were carried out at 37°C as stated in the Methods section.
Dilution of Trizol® LS Inactivated Samples as a Strategy for Viability Testing
One hundred forty microliters of Zika stock virus, a Flaviviridae RG2 virus, was treated with 420 µL of Trizol® LS (3:1 Trizol® LS/virus ratio). A viability test was conducted by adding this 560 µL sample to 560 mL (1:1000 dilution) of media (minimum essential medium with Earl’s salts and L-Glutamine [EMEM] with 10% heat inactivated FBS), and this in turn was added to a HYPERFlask of confluent Vero cells (P1). This was repeated for a total of 3 HYPERFlasks. Flasks were placed into a 37°C incubator. After 48 hours, the media in each flask was completely changed. This step was adopted because it ensured complete recovery of all cells lessening the damage caused by Trizol® LS. This dilution method was compared to removing the Trizol® LS using spin desalting columns (Zeba Spin Desalting columns, Thermo Scientific catalog No. 89893) in which 4 mL of Trizol LS-treated virus (1 mL virus plus 3 mL of Trizol® LS) was applied to the column and eluted in 1 mL of media. This eluted material was added to a single HYPERFlask. Media was not changed in this HYPERFlask. Positive controls consisted of 8 HYPERFlasks, 2 of each infected with approximately 100, 10, 1, and 0.1 pfu per flask. Media in 1 set of each the positive control flasks was also changed at 48 hours; 1 was left untouched. Negative controls (no virus) consisted of 2 HYPERFlasks, 1 infected with media alone and 1 with 560 µL of Trizol® LS diluted in 560 mL of media. After 7 days, 50% of the P1 supernatant infected with ZIKV was passaged to a new set of Vero HYPERFlasks and allowed to propagate again for an additional 7 days (P2). Supernatant from P1 and P2 were then tested in a plaque assay for quantitation of viable virus. Supernatant was diluted 10fold, and 200 µL of each dilution was added to each of well of a 6-well tissue culture plate seeded with Vero cells. A 0.6% agarose overlay was applied and the plates incubated at 37°C and 5% CO2 for 4 days. A secondary overlay with 5% neutral red in 0.6% agarose was added and incubated at 37°C and 5% CO2. One day post staining, the plates were placed on a light box and plaques counted, and the final titers were calculated in PFU per mL (Figures 6 and 7).
Flow chart for dilution of Trizol® LS inactivated samples as a strategy for viability testing. This flow chart demonstrates the inactivation and sample cleanup process for virus stocks inactivated with Trizol LS-based solutions utilizing the dilution method with HYPERFlasks. All incubations were carried out at 37°C as stated in the Methods section.
Flow chart for Trizol® LS inactivation of stock virus and removal of Trizol® LS using desalting columns. This flow chart demonstrates the inactivation and sample cleanup process for virus stocks inactivated with Trizol LS-based solutions utilizing the sample cleanup process with desalting columns. All incubations were carried out at 37°C as stated in the Methods section.
Results
Formalin Inactivation of Cell Monolayers
Cells infected with MP12 virus and treated with neutral buffered formalin for 3.5 hours at 4°C were rendered noninfectious. Cells that were infected with 50 pfu of virus per well (MOI ∼0.00025) resulted in positive viral staining by immunofluorescence (IFA). Immunofluorescence staining results were consistent with previous RVFV assay validation efforts with MOI 4.0 samples resulting in approximately 50% cell infection after 24 hours and assay saturation occurring at MOIs greater that 6.5. 11 Control samples (MOI ∼0.00025) resulted in less than a 1% cell infection rate that was still detectable through IFA. 11 Example of results are shown in Figure 8.
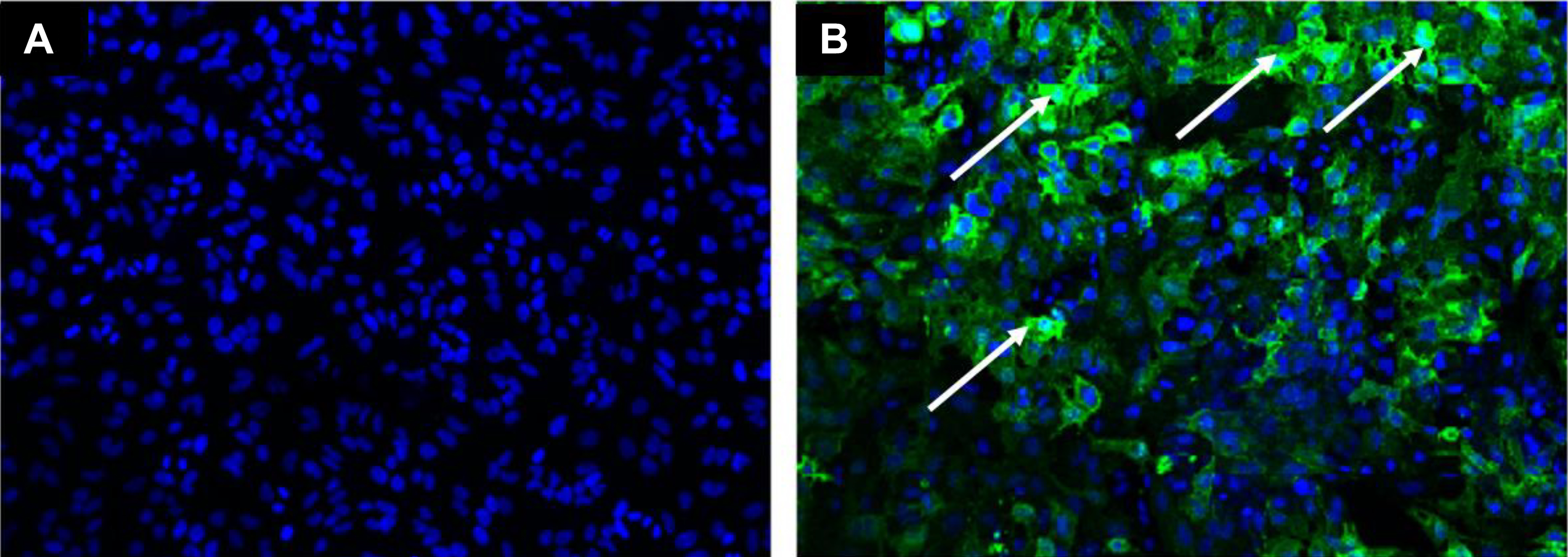
Results from formalin inactivation of MP12 infected cell monolayers through immunofluorescence assay (IFA). IFA results of formalin-treated MP12-infected cell monolayers after 2 passages in a susceptible cell line (A) and a cell monolayer that was spiked with 50 pfu MP 12 virus following filtration to demonstrate cells can still be suitable for viral replication (B). MP12 glycoprotein staining is indicated by the arrows (B).
Formalin Inactivation of Tissues and Removal of Formalin Using Concentrating Columns
Tissues removed from virus infected animals were exposed to formalin for at least 20 days were rendered not infectious. Samples with fixed tissue that were filtered and then had 2000 pfu of virus spiked back into the filter were positive for virus through IFA detection after 2 passages in cell culture, indicating that formalin was removed using the concentrating columns and confirming the capability of the filter to retain virus. Example of results are shown in Figure 9. Cells exposed to non-cleaned-up formalin tissue homogenates exhibited severe cytotoxic effects across both passages and in some cases even during the IFA detection assay. Cells exposed to infected animal tissues that were not treated by formalin solution demonstrated viral propagation following extraction using the concentrating columns. Nonfiltered samples that were spiked with virus were negative for virus, demonstrating the need to mitigate cytotoxic effects of any chemical treatment intended to neutralize virus.
Results from formalin inactivation of virus-infected liver homogenate using immunofluorescence assay (IFA). IFA imaging results of a 20-day formalin-treated virus-infected liver-homogenate-infected cell monolayer after 2 passages in susceptible cells (A) and a cell monolayer that was spiked with 2000 pfu Lassa Josiah following filtration to demonstrate cells can still be suitable for viral replication (B). Virus nucleoprotein is indicated by the arrows (B).
Trizol® LS Inactivation of Stock Virus and Removal of Trizol® LS Using Concentrating Columns
Trizol® LS inactivated all virus infected samples, even at the lowest concentration tested (10%). Results are shown in Figure 10. This is well below the manufacturer’s recommended Trizol® LS concentration for use in RNA extraction. The filter units were fully capable of retaining virus after the samples were diluted, washed to remove Trizol® LS, and then concentrated, despite the high concentration of phenol in Trizol® LS.
Results of Trizol® LS solution inactivation of virus at different concentrations using immunofluorescence assay (IFA). IFA results of concentrated stock virus at 1:9 parts Trizol® LS to sample filtered (A) and at 3:1 parts Trizol® LS to sample after 2 passages in susceptible cell line (B). Part C shows IFA results of 500 pfu of virus that has not been Trizol® LS treated and spiked into the column and washed. Virus glycoprotein staining is indicated by the arrows.
Dilution of Trizol® LS Inactivated Samples as a Strategy for Viability Testing
Trizol® LS is toxic to cells, but when diluted 1:1000 with the media completely changed within 24 to 48 hours after it is initially added to the cell monolayer, the cells overcome the toxicity and can successfully propagate virus. Due to the number of cells in a confluent HYPERFlasks and the volume of media required to keep the monolayer bathed in nutrients, the HYPERFlask is amenable to testing large volumes of inactivated material. As much as 560 µL of Trizol LS-treated virus can be tested in a single flask. The limit of detection for this strategy is approximately 1 pfu per flask after only 1 passage in a susceptible cell line, regardless of whether media is changed 48 hours after infection (Table 1). When used at the recommended concentration, Trizol® LS inactivated 8.05 E+08 pfu/mL of stock virus. This inactivation was demonstrated by both diluting the Trizol LS-treated material 1:1000 and testing the material in a HYPERFlask as well as by removing the Trizol® LS using a desalting column and testing the material without dilution in a HYPERFlask. Results are shown in Tables 1 and 2.
Results from Inactivation of Zika Virus with Trizol® LS Solution and Using HYPERFlasks to Remove Cytotoxic Chemicals.a
Limit of detection results for viability testing using HYPERFlasks with and without completely changing the media 48 hours after infection. MEM is media used in HYPERFlasks; EMEM with 10% heat-inactivated fetal bovine serum. Samples containing 100, 10, 1, or 0.1 plaque forming unit (pfu) of virus was added to 56 0mL of media and added to a HYPERFlask. Negative control shown consisted of only media (no virus) added to a HYPERFlask. Cytopathic effect (CPE) was minimal after the first passage, but evident with 100% of the cells involved after the second passage. The amount of viable virus, or titer, after 7 days in culture was quantitated using a plaque assay. Well over 6 logs of virus were recovered after only 1 passage in Vero cells. Changing the media on day 2 did not affect the ability to recover virus. N.D., no virus detected.
Results from Inactivation of Zika Virus with Trizol® LS Solution and Using Desalting Columns to Remove Cytotoxic Chemicals.a
Results of viability testing of Trizol LS-treated stock virus in HYPERFlasks, after diluting 1:1000 and completely changing the media 48 hours after infection as compared to removing the Trizol® LS using a ZEBA desalting column and applying the entire sample to a HYPERFlask® without changing the media. MEM is media used in the HYPERFlasks; EMEM with 10% heat-inactivated fetal bovine serum. Samples (0.56 mL) of Trizol LS-treated virus was added to 560 mL of media (1:1000 dilution) and added to a HYPERFlask. Negative control shown consisted of only media (no virus) added to a HYPERFlask. Cytopathic effect (CPE) was not noted in any passage. The amount of viable virus, or titer, was quantitated using a plaque assay. No viable virus was recovered from either passage of Trizol LS-treated stock virus. N.D. indicates no virus detected.
Discussion
Various means of analyzing whether a sample has been rendered noninfectious have been used to varying degrees of sensitivity.4-5,13 Analyzing inactivation in the presence of particularly cytotoxic chemicals can be challenging because any amount of infectious virus would still render the sample a Select Agent and pose as a public health risk, particularly when samples are downgraded in their biosafety requirements and no longer closely monitored through inventory control processes. The incorporation of 2 passages of the inactivated material alongside samples also containing trace amounts of virus more clearly resolves whether all infectious material has been inactivated.
All inactivation testing demonstrates that both formalin and Trizol® LS solutions successfully inactivate virus infected cell lines and tissues, which is consistent with previously published literature.5 -16 Our objective in these studies was to demonstrate a series of methods that can be used to remove cytotoxic chemicals to allow research institutes working with Select Agents to comply with the latest regulations while minimizing the amount of resources needed to develop cleanup methods from scratch. In our methods, we pursued a series of strategies, the use of concentrating columns or desalting columns for removal of cytotoxic chemicals and retention of inactivated virus or HYPERFlasks that dilute any cytotoxic chemicals sufficiently to avoid degradation of the cell lines.
Each of these methods has its benefits and trade-offs that must be evaluated by entities to best fit their research needs. Desalting columns can be used with the largest sample volumes (1 mL/column) but can easily plug when used for complex matrices. Because of this, their use would not be recommended for entities evaluating efficacy of cytotoxic chemicals against infected samples of whole blood or tissue specimens. Concentrating columns are less susceptible to plugging compared to the desalting columns, but sample volumes are limited to less than 100 µL per column because the sample must be diluted 100 times prior to column concentration. This level of dilution is required to minimize degradation of the column filter from any cytotoxic chemicals. Dilution through the use of HYPERFlasks increases the sample volume to approximately 140 µL but is a more labor-intensive process due to the requirement of a 48-hour media change to eliminate cytotoxicity issues. However, this method is not limited by sample complexity, so it is suitable for either whole blood or tissue homogenates.
Efficacy of the viral inactivation procedure was confirmed either through a plaque assay or IFA following 2 cell passages requiring at least 50% of cell culture retained between passages. Our institution has traditionally relied on 2 consecutive passages in viral cell culture to maximize potential for detecting low levels of residual live virus. Previous validated methods that were not subjected to this same standard of cell passaging have later been shown to be ineffective.4,13,14 Due to this history, it is our recommendation that any method demonstrating inactivation of virus should be validated using a viability test that utilizes 2 consecutive passages in cell culture prior to any detection assay.
Each of these studies demonstrated successful inactivation of a variety of viruses with either Trizol® LS or formalin solutions, which when combined with previous literature and a recent publication of formalin efficacy against Tier 1 Select Agent bacteria demonstrates that these are both highly effective and reliable methods to inactivate Select Agents.15,16 While sample cleanup and detection methods for viruses were optimized through these procedures, the studies presented do not demonstrate any new evidence of efficacy of formalin or Trizol-based solutions against viruses. In addition, our research institute had to absorb costs both in terms of supplies and personnel hours to reach these conclusions, adding up to over $200 000 for each of the assays. As an example, a single 500-µL sample inactivated using Trizol® LS solution followed by dilution using HYPERFlasks and detection through a plaque assay would cost approximately $3000, not including personnel hours or non-cell-culture-related supplies and materials, including personal protective equipment. This type of cost for inactivation and viability testing of a single sample of virus presents a very resource-intensive requirement for select agent laboratories. Based on our results along with those of our colleagues, it is recommended that the regulatory authorities reevaluate the requirements for in-house validation of well-established inactivation methods. In addition, regulatory authorities should also consider developing a bank of well-established methods such as formalin- and Trizol-related inactivation methods that should be exempt from the in-house validation requirement. The use of an inactivation method bank would add balance to the regulations by maintaining the need for in-house testing of novel or less established methods while also reducing the resource and administrative burden on entities using procedures that have been previously validated.
Footnotes
Authors’ Note
The views, opinions, and/or findings contained herein are those of the authors and should not be construed as an official Department of Army position, policy, or decision unless so designated by other documentation. This work was presented at the 2017 Chesapeake Area Biosafety Association Annual Symposium on June 14, 2017, in Frederick, MD; the MidWest Area Biosafety Network Symposium on August 8, 2017, in Iowa City, IA; and the American Biosafety Association International Meeting on October 16, 2017, in Albuquerque, NM.
Acknowledgments
We are indebted to Ms Cindy Rossi, who provided contributions to the content in the manuscript as well as technical assistance with protocols.
Ethical Approval Statement
Not applicable to this study.
Statement of Human and Animal Rights
Not applicable to this study.
Statement of Informed Consent
Not applicable to this study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Defense Threat Reduction Agency and The Department of the Army.