
Abstract
There is a common assumption that viral lysis buffers are sufficient to render viruses noninfectious. This assumption has a significant impact on the way biological samples are processed, labeled, and handled for shipment. Several lysis buffers, including TRIzol, AVL, RLT, MagMAX, and easyMAG, were examined for their capacity to inactivate representative viruses from multiple genera, including alphavirus, bunyavirus, flavivirus, adenovirus, enterovirus, influenza B, and simplexvirus. Viruses were noninfectious following treatment with TRIzol, MagMAX, and easyMAG buffers, while some viruses were still viable in cell cultures following treatment with AVL and RLT buffers. These results indicate the need to further evaluate the expectation that lysis buffers render live viruses inactive, allowing handling and processing of these samples under low-level containment conditions.
Most methods to extract viral nucleic acids use buffers that contain chaotropic agents, such as guanidine thiocyanate (GITC) or other protein denaturants. There is a general assumption that the denaturing activity of these agents is sufficient to inactivate all viruses. Reliance upon this assumption may provide a false sense of security in the subsequent handling and processing of lysed specimens. Furthermore, viral titers of stocks used for clinical or experimental testing are highly variable, and therefore, the ability to achieve inactivation may also vary. Given that incomplete inactivation of viruses has important safety implications, commonly used buffers must be validated for their ability to render a virus inactive.
Our organization comprises different laboratories handling various viral specimens according to each laboratory’s unique expertise. Depending on the specimen and test, samples are often transported to different laboratories for further analysis. Since different facilities operate under different biosafety levels and viruses that undergo lysis are often transported as “noninfectious,” confirmation of inactivation of viral samples is necessary to ensure personnel safety. In this study, we examined several lysis buffers for their ability to render suspensions of viruses noninfectious to cell culture.
Materials and Methods
Lysis buffers tested in the study were TRIzol Reagent (Invitrogen, Carlsbad, California), AVL buffer (Qiagen, Valencia, California), RLT buffer (Qiagen), MagMAX 96 buffer (Thermo Fisher, Waltham, Massachusetts), and easyMAG buffer (bioMerieux, Hazelwood, Missouri).
A representative panel of viruses was chosen from 7 different genera routinely used by 2 laboratories participating in this study. The following viruses were tested: Eastern equine encephalitis virus (EEEV; Togaviridae, Alphavirus), Chikungunya virus (CHIKV; Togaviridae, Alphavirus), West Nile virus (WNV; Flaviviridae, Flavivirus), La Crosse virus (LACV; Bunyaviridae, Bunyavirus), adenovirus type 4 (AdV4; Adenoviridae, Mastadenovirus), echovirus type 6 (ECHO; Picornaviridae, Enterovirus), influenza type B (FLUVB; Orthomyxoviridae, Influenzavirus B), and herpes simplex virus type 1 (HSV-1; Herpesviridae, Simplexvirus). Viral work was done in biosafety level 3 for EEEV, CHIKV, WNV, and LACV and biosafety level 2 for AdV4, ECHO, FLUVB, and HSV-1. Sources and strains of viruses are shown in Table 1.
List of Viral Strains.
TRIzol, AVL, RLT, and MagMAX buffers were evaluated with EEEV, CHIKV, WNV, and LACV, while easyMAG buffer was evaluated with AdV4, ECHO, FLUVB, and HSV-1. WNV, EEEV, CHIKV, and LACV were evaluated at the New York State (NYS) Arbovirus Laboratory, while AdV4, ECHO, FLUVB, and HSV-1, all derived from clinical isolates amplified in-house, were evaluated at the NYS Clinical Virology Laboratory. Tubes were prepared separately with viruses and buffers. Lysis and subsequent testing of infectiousness were completed only for buffer-virus pairs using an established methodology in each laboratory, and therefore, different viruses were subjected to different treatments. Viral titers were calculated as plaque-forming units (PFU) per milliliter for all viruses tested.
Lysis conditions were performed as follows: briefly, 140 μL WNV (8.5 log10 PFU/mL), CHIKV (7.7 log10 PFU/mL), and LACV (5.4 log10 PFU/mL) were mixed with 560 μL AVL. Then, 100 μL of the same viruses was added to 350 μL RLT and 1000 μL TRIzol, respectively. For the MagMAX lysis buffer, 50 μL EEEV (8.1 log10 PFU/mL), WNV, and LACV was added to 100 μL buffer. All virus-buffer mixtures were held at room temperature for 10 minutes, and a separate 10-minute additional incubation at 65°C was performed on a subset of samples for WNV and CHIKV in both AVL and RLT buffers. Thereafter, a 100-fold dilution in Hank’s balanced salt solution (BA-1) was prepared prior to infections based on preliminary experimentation assessing cytotoxicity of lysis buffers on cell culture (data not shown). Viral stocks of WNV, CHIKV, EEEV, and LACV, also diluted in the same manner but without lysis buffer, served as positive controls.
Confluent African green monkey kidney cells (Vero, ATCC CCL-81) in 6-well plates were inoculated in duplicate wells with 200 μL diluted virus/buffer or controls. Virus absorption was carried out at 37°C in an incubation chamber (Thermo Fisher) with 5% CO2 for 1 hour, after which 3 mL minimal essential medium (MEM) supplemented with 2% fetal bovine serum (FBS) was added to each well. Cytopathic effect (CPE), the visible structural changes in the host cell due to virus replication and amplification, was used to determine viability of WNV, LACV, CHIKV, and EEEV in this experiment. Log10 reduction of viral titers was calculated based on the assumption that a single infectious particle is sufficient to establish cytopathology. Dilutions of the virus to avoid cell toxicity were factored into the final calculations. Plates were examined daily for CPE for 1 week, and negatives were passaged once a week for 3 weeks. Final assessments were made at week 5.
For AdV4 (7.5 log10 PFU/mL), ECHO (6.5 log10 PFU/mL), FLUVB (6.5 log10 PFU/mL), and HSV-1 (7.0 log10 PFU/mL), 100 μL of each isolate was added to 500 μL of the easyMAG lysis buffer and held at room temperature for 10 minutes. Treated virus suspensions were immediately diluted 500-fold in 0.05% gelatin in Tris–Hank’s balanced salt solution prior to inoculation, based on preliminary experiments assessing the cytotoxicity of the lysis buffer on cell culture (data not shown). Aliquots from the same stocks of AdV4, ECHO, FLUVB, and HSV-1 were diluted in the same manner but without prior lysis buffer treatment and inoculated as positive (untreated) controls. Then, 300 μL of each diluted lysis buffer-treated virus or untreated controls was inoculated into glass culture tubes (16 × 125 mm) containing monolayers of cell lines known to be highly permissive for infection and replication of the viruses. Adenocarcinoma human alveolar basal epithelial cells (A549, ATCC CCL-185), human rhabdomyosarcoma cells (RD, ATCC CCL-136), human embryonic lung cells (HEL, ATCC CCL-137), and Madin-Darby canine kidney cells (MDCK, ATCC CCL-34) were inoculated with AdV4, ECHO, HSV-1, and FLUVB, respectively; allowed to absorb for 1 hour at 37°C with 5% CO2; and replaced with Glasgow’s minimal essential media (GMEM) supplemented with 2% FBS. Microscopic observation of the inoculated monolayers was monitored for the development of CPE 3 times per week, and cells were harvested for immunofluorescent staining when CPE was covering more than 25% of the monolayer. CPE-negative cultures were harvested at day 10 for HSV-1 and at day 14 for AdV4, ECHO, and FLUVB. All harvests were immunostained to confirm the presence or absence of virus growth, with primary antibodies for AdV4, ECHO, HSV-1, and FLUVB from EMD Millipore (Billerica, MA), Quidel Diagnostic Hybrids (San Diego, CA), Trinity Biotech (Jamestown, NY), and EMD Millipore, respectively. Cells were trypsinized off the glass tubes, and 4-well slides of harvested cells were prepared from cultures. Indirect immunofluorescence assays (IFAs) were used to detect virus growth according to the manufacturers’ instructions. Briefly, a 30-minute incubation with primary antibody was followed by 2 washes, and then fluorescein isothiocyanate–conjugated secondary antibody was added for an additional 30 minutes followed by several washes. Stained cells were visualized with fluorescence microscopy. All experiments were performed 3 times to ensure reproducibility of results.
Results and Discussion
Control virus infections of AdV4, ECHO, HSV-1, and FLUVB that had not been treated with lysis buffer prior to inoculation developed CPE to 25% or more of the monolayer by days 6, 6, 5, and 8 post-inoculation, respectively. Cell harvests from these cultures all produced positive specific fluorescence on virus antigen IFA tests. All viruses treated with easyMAG buffer failed to produce CPE in culture (Table 2), and no foci were detected by IFA on any of the cell harvests. These viruses were inoculated at the highest possible titers, given the available virus stocks and the dilutions needed to overcome cell toxicity of the lysis buffer. The easyMAG buffer inactivated all viruses tested, corresponding to an equal to or greater than 4.5, 3.5, 3.5, and 4.0 log10 PFU reduction in titers of AdV4, ECHO, FLUVB, and HSV-1, respectively (Table 2).
Viral Titers and Log10 Reduction of Viruses Tested.
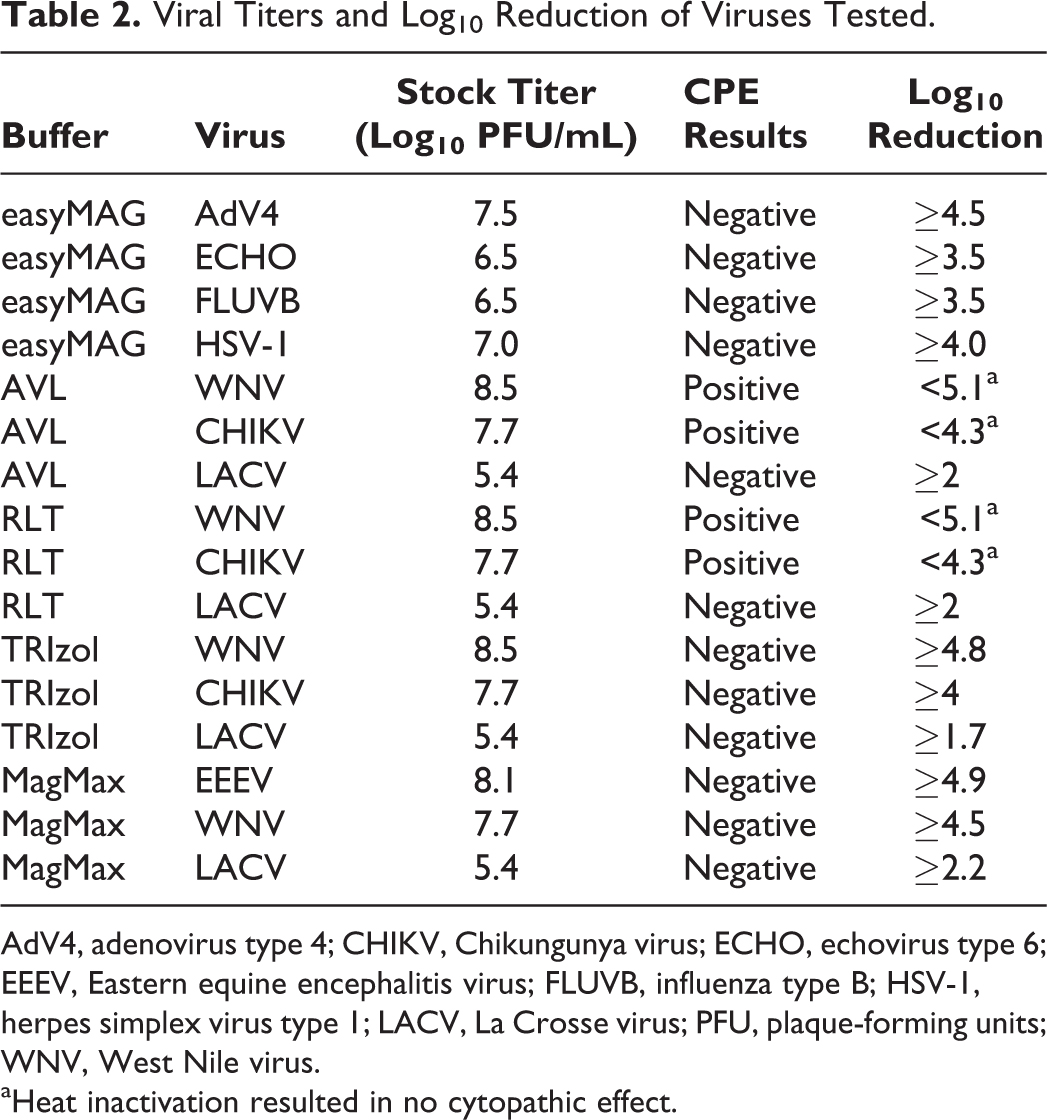
AdV4, adenovirus type 4; CHIKV, Chikungunya virus; ECHO, echovirus type 6; EEEV, Eastern equine encephalitis virus; FLUVB, influenza type B; HSV-1, herpes simplex virus type 1; LACV, La Crosse virus; PFU, plaque-forming units; WNV, West Nile virus.
Heat inactivation resulted in no cytopathic effect.
CPE was not detected in Vero cell culture following treatment of viruses with TRIzol or MagMAX reagents. In contrast, CPE was observed in cells inoculated with WNV and CHIKV treated with RLT and AVL buffers (Table 2). This could be due to different denaturing ingredients and/or their concentrations in TRIzol and MagMAX buffers. A previous study using multiple viruses, including EEEV and WNV, determined that AVL buffer was sufficient for virus inactivation. 1 We used CHIKV with AVL in our study, and intrinsic susceptibility of viruses to chemical inactivation can vary. The concentration of WNV was higher in our experiments, which could account for the residual infectivity. However, Smither et al 2 reported that buffer AVL alone does not inactivate Ebola virus and that a separate heat step is required. Similar findings of insufficient inactivation of virus with AVL and RLT buffers alone were reported by Haddock et al, 3 which corroborates our work. This provides further evidence that lysis buffers should be individually assessed using laboratory viral stocks and that standard procedures for viral inactivation should be reevaluated.
Residual infectivity can be seen with both WNV and CHIKV but not with LACV. This could be due to differences either in susceptibility to the lysis buffers or in the high starting titers. Since infectivity was detected with these 2 viruses, a separate 10-minute incubation in lysis buffer at 65°C was performed. The disruptive effect of heat incubation on viral envelopes and genomes is well documented,4,5 and as expected, infectivity was not observed when the heat-treated virus-buffer mixtures were subsequently cultured and passaged. These results suggest that an additional heat step should be considered when using these buffers if inactivation is not initially achieved.
Measurement of inactivation is limited by the technical aspects of each assay. Lysis buffers/viral mixtures must be diluted to avoid toxicity in cell-based assays for virus titers; therefore, fold reduction is always less than the starting viral titers. Caution should be taken when inactivating viruses at titers higher than the validated fold reduction.
The ability of lysis buffers to render viruses noninfectious is essential to ensure proper handling and facilitate performing subsequent experimentation and testing at lower biosafety levels. Rendering a virus noninfectious can be difficult, as even fixatives such as acetone or 4% formaldehyde have been shown to be inconsistent in their capacity to fully inactivate viruses.6,7 Numerous cases of laboratory-acquired infections from improperly inactivated or mishandled infectious agents have been reported,8 –10 and there are significant risks that come with improper transport and dissemination if infectious materials are not properly labeled or packaged. In addition to the obvious health risks, institutional repercussions can significantly jeopardize both the individual laboratory and the greater research community.
Laboratories often rely on the assumption that standard protocols for viral inactivation are universal, yet studies support the need to further assess frequently used methods using individual viruses and experimental conditions. For example, standard plaque neutralization assay requires a heat inactivation step of 56°C for 30 minutes, yet this procedure for viral inactivation has shown to be ineffective against certain alphaviruses.11,12 Also, viral storage media containing 20% protein can enhance the thermostability of viruses such as severe acute respiratory syndrome coronavirus. 13
We have compared the ability of multiple commonly used lysis buffers, AVL, RLT, MagMAX, TRIzol, and easyMAG, in their ability to render various viruses noninfectious. Taken together, this report underscores the significance of assessing residual infectivity of “inactivated” agents in individual laboratories and being cognizant of the limitations of each specific assay in determining whether an agent is fully inactivated.
Footnotes
Acknowledgments
We thank the New York State Department of Health, Wadsworth Center Tissue Core for providing cell cultures and the Media Department for their media services.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.