
Abstract
Prior to the release from BSL-3 or -4 laboratories, materials from which nucleic acids are to be extracted must be rendered noninfectious. Typically, phenol/guanidine thiocyanate reagents such as TRIzol LS have been used to extract and inactivate RNA from infectious viruses. Because of the toxicity of this reagent to the tissue culture cells used to propagate viruses, it has been difficult to fully evaluate the sterility of extracted viral RNA using tissue culture. In this study, we have demonstrated the use of sucrose cushion centrifugation to remove TRIzol LS from extracted viral suspensions. The inactivated viral suspensions were then sterility tested without confounding cell toxicity from TRIzol LS. This method allowed a clear demonstration of the use of TRIzol LS to inactivate a broad group of viral families that include Togaviridae, Arenaviridae, Bunyaviridae, Coronaviridae, Filoviridae, Flaviviridae, and Paramyxoviridae at titers ranging from a high of 106.1 to 108.3 tissue culture infectious dose 50% (TCID50/mL) to the lower limit of virus detection in cell culture. TRIzol LS used in a ratio of 4 parts TRIzol LS to 1 part viral suspension inactivated representative viruses of all families tested.
Introduction
Many diagnostic assays such as polymerase chain reaction (PCR) and advanced molecular biology applications that include whole genome sequencing can be performed on extracted and noninfectious viral RNA, enabling analyses to be conducted at lower levels of biocontainment as well as reducing the risk of infection of laboratory personnel by allowing them to work outside of the containment laboratory.
Phenol/guanidine thiocyanate reagents, such as TRIzol LS, are useful for the extraction and inactivation of viral nucleic acids. However, their ability to eliminate infectious material in an extracted sample can be difficult to assess since phenol/guanidine thiocyanate is toxic to cell culture even at dilutions 1:100 of standard use concentrations and below.1 –3 To circumvent the cell culture toxicity and difficulty to evaluate sterility, studies have separated phenol/guanidine thiocyanate from aqueous RNA and possible virus-containing solutions by dialysis or through the use of sucrose cushion centrifugation prior to sterility testing in cell culture.4,5 These studies have utilized Zaire ebolavirus as a representative virus for all enveloped, negative-, or ambi-sense RNA viruses.
Towner et al 5 have used a 20% to 60% sucrose cushion centrifugation procedure to separate a phenol/guanidine thiocyanate reagent from aqueous RNA and possible virus-containing solutions. They would collect virus at the 20% to 60% sucrose interface with virus recoveries generally >50%. We have modified their procedure by deleting the 60% sucrose cushion and collecting virus following centrifugation through a 20% sucrose cushion. This modification increased the sensitivity of sterility testing by enabling virus recoveries of generally >80% and allowed the evaluation of TRIzol LS for its ability to inactivate representative viral species for a range of viral families, including Togaviridae, Arenaviridae, Bunyaviridae, Coronaviridae, Filoviridae, Flaviviridae, and Paramyxoviridae.
Materials and Methods
Virus Propagation
All viruses were propagated in Vero cells obtained from ATCC (CCL81). Eighty percent to 95% confluent cell cultures were inoculated at a multiplicity of infection (MOI) of 0.001 and propagated at 37°C, 5% CO2 in growth medium consisting of Glasgow Minimal Essential Media (GMEM) supplemented with 10% fetal bovine serum and 1% antibiotic-anti-mycotic (ThermoFisher Scientific product No. 15240-062). Culture supernatant was collected 6 to 10 days post-inoculation, clarified via centrifugation at 2000 × g for 10 minutes, and used as virus preparation for RNA extraction and inactivation.
Virus Inactivation by TRIzol LS
Virus preparations (Table 2) were inactivated in TRIzol LS (ThermoFisher Scientific product No. 10296-010) by adding 140 µL of virus into 560 µL of TRIzol LS Reagent (1:4 ratio), mixing, and then incubating at room temperature for 10 minutes.
Immunofluorescence Assay Reagents.
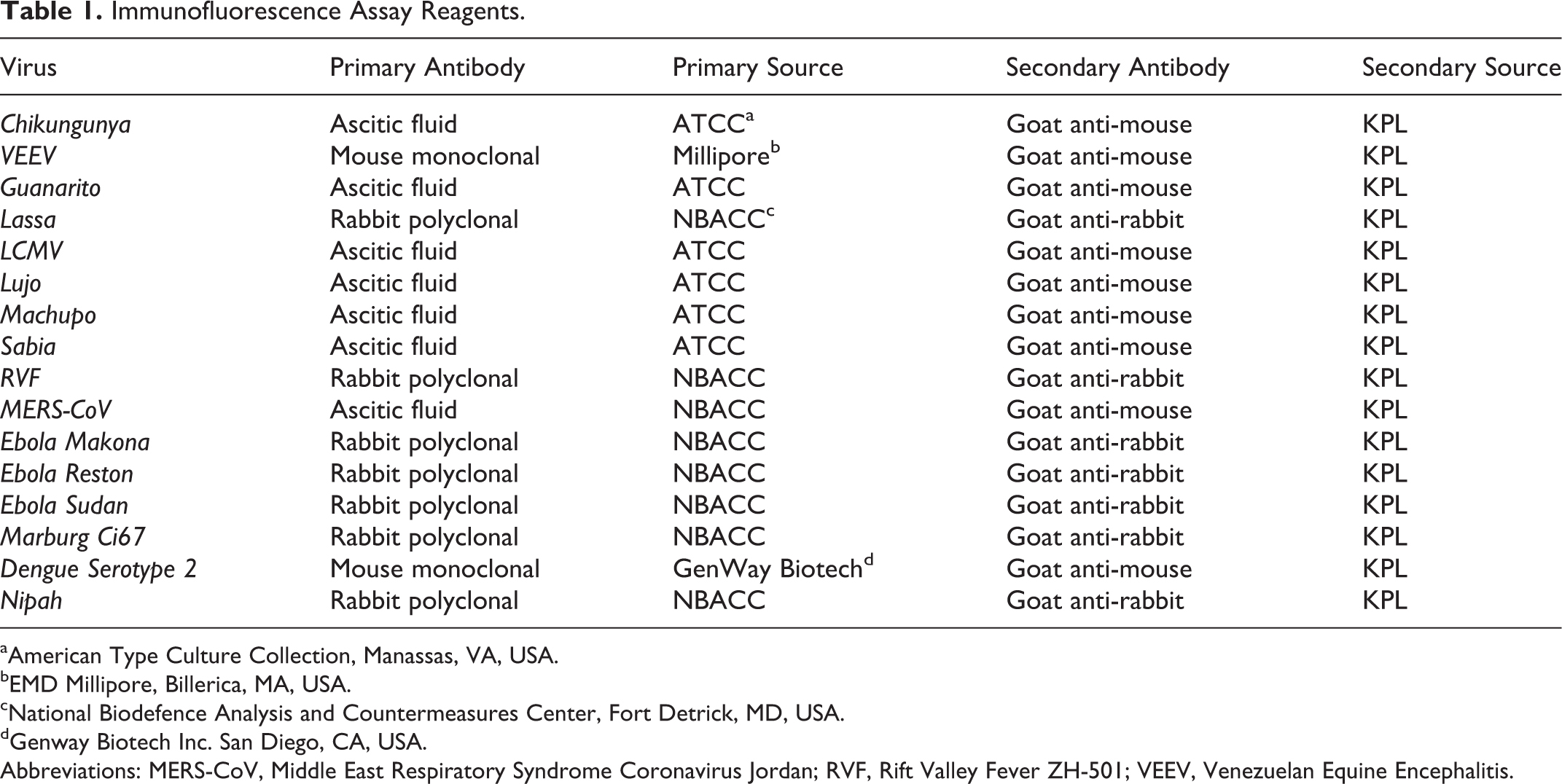
American Type Culture Collection, Manassas, VA, USA.
EMD Millipore, Billerica, MA, USA.
National Biodefence Analysis and Countermeasures Center, Fort Detrick, MD, USA.
Genway Biotech Inc. San Diego, CA, USA.
Abbreviations: MERS-CoV, Middle East Respiratory Syndrome Coronavirus Jordan; RVF, Rift Valley Fever ZH-501; VEEV, Venezuelan Equine Encephalitis.
TRIzol LS Inactivation of Viruses.a
Abbreviations: MERS-CoV, Middle East Respiratory Syndrome Coronavirus Jordan; RVF, Rift Valley Fever ZH-501; VEEV, Venezuelan Equine Encephalitis.
In triplicate, TRIzol LS treated and mock-treated virus samples were centrifuged through sucrose, and viable virus was visualized by CPE and IFA. Mock-treated viral samples were quantitated by TCID50. N/A No CPE in Vero cells.
Results from 3 replicate experiments.
Sucrose Cushion Preparation and Ultracentrifugation
A 20% weight per volume sucrose solution was created by adding sucrose (Sigma-Aldrich product No. S1888-500G) into Hanks Balanced Salt Solution (HBSS, Sigma Aldrich product No. 14025-092) and then vortex mixing for 1 minute. Ten milliliters of sucrose solution was added to 38.5 mL capacity ultracentrifuge tubes (Beckman Coulter product No. 344058). Twenty milliliters of phosphate buffer solution (PBS, ThermoFisher product No. 10010-072) were gently layered on top of the sucrose solution. If a visible gradient between the 20% sucrose solution and PBS was not formed, a new tube was obtained and the process repeated. One hundred forty microliters of live virus or 700 µL of inactivated material was then added followed by additional PBS until liquid was within 5 mm of the top of the tube per manufacturer’s instructions. Samples were placed in a SW32Ti Rotor and centrifuged at 25 000 RPM for 2 hours in a Beckman Coulter Optima L-100XP Ultracentrifuge to pellet virus particles while leaving TRIzol LS in the PBS. After centrifugation, all liquid was carefully aspirated, and the pelleted material was rehydrated in 2 mL of GMEM.
Immunofluorescence Assay
All virus samples treated with TRIzol LS were tested for viability by culture on Vero cells followed by indirect fluorescent antibody assay (IFA). 6 Two hundred microliters from each resuspended pellet were added to a T-25 cell culture flask containing Vero cells at 80% to 90% confluency and GMEM. The cultures were then incubated at 37°C and 5% CO2 for 10 days. On day 10, the flasks were scraped and their contents aspirated into conical vials. The aforementioned virus isolation procedure was repeated using 0.5 mL of the harvested material from the first T25 culture for another 10 days and observed for cytopathic effects (CPE). The cell slurries from both passages were applied to slides following the method of Hsiung. 7 Droplets of cell suspension were applied to slides and allowed to dry at room temperature in a biosafety cabinet. The sides were fixed for 15 minutes in acetone to permeabilize the cell membrane and secure the cells to the slide. After acetone fixation, the slides were stained with an antivirus antibody specific for the virus being tested, followed by a fluorescein-labeled goat anti-species antibody (Table 1). Slides were then microscopically screened to detect cells exhibiting fluorescence. Negative control slides were prepared for each virus tested by inoculating Vero cell cultures with 200 µL GMEM and processing them, as previously described, for IFA. Positive control slides were prepared by inoculating 103 tissue culture infectious dose 50% (TCID50) of each virus tested into Vero cell cultures and processing them, as previously described, for IFA.
Tissue Culture Infectious Dose 50%
A TCID50 assay was adapted from Lennette 8 for quantitating viable virus. Two hundred and twenty-two µL aliquots from each resuspended viral pellet were placed into wells of a 96-well plate containing Vero cells at 95% confluency. The aliquots were serially diluted 1:10 in the plates for a total of 11 dilutions and incubated for 1 hour at 37°C and 5% CO2. The wells filled with undiluted inoculum were then aspirated, and fresh GMEM was added. The plates were then placed back into 37°C and 5% CO2 for 10 days. For CPE-causing viruses, cell monolayers were observed microscopically for the presence of CPE. The TCID50 endpoint titer was calculated following the method of Reed and Meunch. 9 Negative control plates were prepared for each virus tested by inoculating Vero cell cultures with 200 µL GMEM and processing them, as previously described, for CPE. Positive control plates were prepared by inoculating 103 TCID50 of each virus tested into Vero cell cultures and processing them, as previously described, for CPE.
Lymphocytic choriomeningitis (LCMV), Machupo, and Lujo viruses did not cause readily observable CPE in Vero cell monolayers. TCID50 determinations were made, following the calculation method of Reed and Muench, 9 by monitoring 10-day cultures of serial sample dilutions for virus viability by IFA.
Sucrose Cushion Process Loss
To determine the viral loss of separating virus from TRIzol LS by centrifugation through a sucrose cushion, 140 µL of live virus was centrifuged through a 20% sucrose cushion. The TCID50 of the collected virus was subtracted from the TCID50 of the input virus. The differences were expressed in log10. All reactions were performed in triplicate, and virus recoveries were expressed as the mean virus recovery. Similarly, the process loss was expressed as mean log10 ± SD.
Results
Alphaviruses (chikungunya [Chik], Venezuelan equine encephalomyelitis [VEE]), Arenaviruses (Guanarito, Lassa, LCMV, Lujo, Machupo, Sabia), a Bunyavirus (Rift Valley fever [RVF]), a Coronavirus (Middle East respiratory syndrome coronavirus [(MERS-CoV]), Filoviruses (Ebola Makona, Ebola Reston, Ebola Sudan, Marburg Ci67), a Flavivirus (Dengue serotype 2), and a Paramyxovirus (Nipah) were selected for the evaluation of the inactivation of viruses with TRIzol LS. Viral stocks used for these experiments were quantitated by TCID50 to ascertain input viable virus numbers, and mock-treated viral samples were quantitated by TCID50 to estimate process loss, which is the amount of assay input virus minus the amount of recovered virus (Table 2). The process loss ranged from 0 log10 for LCMV to 0.47 log10 for MERS-CoV. For the majority of the viruses tested, the process loss was 0.10 to 0.20 log10 of input virus.
TRIzol LS–treated virus samples were centrifuged through the sucrose cushion, and viable virus was quantitated by CPE and IFA. For those viruses that do not produce CPE, virus was quantitated by IFA only. For virus suspensions at titers of 106.1 to 108.3 TCID50/mL, no viable virus could be recovered from viruses treated with TRIzol LS as visualized by CPE and IFA. For virus suspensions of titers of 106.2 to 107.0 TCID50/mL that do not form CPE in Vero cells, no virus viability was detected by IFA (Table 2) following treatment with TRIzol LS.
Discussion
In this study, TRIzol LS extraction effectively inactivated all Alpha, Arena, Bunya, Corona, Filo, and Paramyxo viruses examined to the limits of detection of the utilized virus viability assays. The sterility assay limit of detection was the process loss of the sucrose cushion purification method. For most viruses examined in this study, the process loss was generally 0.1 to 0.2 logs of the input virus, which was consistent with virus titer loss from manipulations such as freeze thawing, purification, and centrifugation observed in other studies.10 –12
Variables in this study were the virus:TRIzol LS ratio (vol:vol) and the type of virus preparations that were used. The virus:TRIzol LS ratio was 1:4, which was more stringent than the 1:3 recommended by the manufacture and the virus preparations were clarified culture supernatants. If other reaction conditions are utilized such as different virus:TRIzol LS ratios, types of virus preparations such as cell lysates, or other viruses, it is suggested that the sterility of the Trizol LS extracted and purified RNA be verified following the methodology of this study prior to release from biocontainment.
Footnotes
Authors’ Note
The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the DHS or S&T. In no event shall DHS, NBACC, S&T, or Battelle National Biodefense Institute have any responsibility or liability for any use, misuse, inability to use, or reliance upon the information contained herein. DHS does not endorse any products or commercial services mentioned in this publication.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded under Contract No. HSHQDC-15-C-00064 awarded by the Department of Homeland Security (DHS) Science and Technology Directorate (S&T) for the operation and management of the National Biodefense Analysis and Countermeasures Center (NBACC), a Federally Funded Research and Development Center.