
Abstract
In this study, the antimetastatic potential of the ethanolic extract of Aerva lanata was evaluated using the B16F-10 melanoma–induced lung metastasis model. Metastasis was induced in C57BL/6 mice by injecting highly metastatic B16F-10 melanoma cells through the lateral tail vein. Simultaneous treatment with A lanata inhibited tumor nodule formation in the lungs (70.53%), and there was a 65.3% increase in the survival rate of metastatic tumor–bearing animals. These results correlated with biochemical parameters such as lung collagen hydroxyproline, hexosamine, and uronic acid contents; serum sialic acid and γ-glutamyl transpeptidase levels; and histopathological analysis. In vitro studies using B16F-10 cells showed that A lanata inhibited migration of tumor cells, cell invasion through type-I collagen-coated polycarbonate filter and activation of matrix metalloproteinases. Treatment with A lanata induced apoptotic response, characterized by apoptotic morphology, a typical ladder of DNA fragmentation, and detection of 3′ hydroxyl ends in DNA by TUNEL assay. There was an increase in the percentage of cells in the sub-G0/G1 phase indicating cell cycle arrest. A lanata treatment resulted in downregulation of bcl-2 and cyclin-D1 expression and upregulation of p53, bax, caspase-9, caspase-3, p21, and p27 gene expression in B16F-10 cells. Proinflammatory cytokine production and gene expression were also found to be downregulated in A lanata–treated cells.
Introduction
Cancer is one of the leading causes of death worldwide. Among cancers, melanoma is the most malignant skin cancer, and its occurrence has remarkably increased during the past few decades. 1 Worldwide, 9 million deaths result from cancer each year, which may rise to 20 million by 2020, as anticipated by WHO.
The hallmarks of a cancer cell are self-sufficiency of growth signals, resistance to growth-inhibitory signals, resistance to apoptosis, sustained angiogenesis, and the acquisition of invasive and metastatic potential. Most malignancies have defects in the apoptotic regulatory pathways, leading to defects in apoptosis. So one of the crucial therapeutic strategies against cancer is to trigger a cellular suicide pathway and induce tumor cell apoptosis in response to stimuli using suitable drugs. 2
The most devastating aspect of cancer is metastasis, which is often resistant to conventional therapies such as chemotherapy and radiation therapy. In the majority of cancer patients, by the time of diagnosis of the primary tumor, metastasis to the regional lymph node and/or distant organs has occurred. 3 An important step in metastasis is the breakdown of connective tissue barriers, which is mediated by proteolytic activity. Typically, proteases such as matrix metalloproteinases (MMPs) are overexpressed and are localized to the tumor cell surface at the invadopodia to mediate extracellular matrix (ECM) breakdown and facilitate invasion. 4
Apoptosis is a controlled physiological process that occurs in a morphologically and biochemically distinct manner and ultimately leads to cell death. The process involves a sequence of events, including cell shrinkage, increased cytoplasmic density, chromatic condensation and segregation into sharply circumscribed masses, and the formation of membrane-bound surface apoptotic bodies. 5 Apoptotic cells are phagocytized from the midst of living tissues by neighboring cells or macrophages without eliciting an inflammatory reaction. The apoptotic process is misregulated in various diseases, including cancer, and alterations in the function of apoptosis-related genes have a profound influence on tumor progression. Therefore, activation of the apoptotic pathway in tumor cells is considered to be a protective mechanism against the development and progression of cancer.
Aerva lanata, belonging to the family Amaranthaceae, is an important medicinal plant grown as a common garden plant in India. It is used in Indian folk medicine for the treatment of diabetes mellitus, urinary calculi, hematemesis, bronchitis, nasal bleeding, cough, and so on. 6 Our previous studies with ethanolic extract of A lanata showed that it could enhance the total white blood cell count, the number of plaque-forming cells in the spleen, circulating antibody titer, and proliferation of splenocytes, thymocytes, and bone marrow cells and inhibit Dalton’s lymphoma ascites (DLA)-induced solid tumor development in mice. 7 In the present study, the effect of ethanolic extract of A lanata on the inhibition of pulmonary metastasis induced by B16F-10 melanoma cells in C57BL/6 mice was evaluated with special emphasis on the mechanism of action, using in vitro models.
Materials and Methods
Animals
C57BL/6 mice (20-25 g body weight, 6- to 8-week-old males) were purchased from the National Institute of Nutrition, Hyderabad, India. The animals were fed with mouse chow (Sai Feeds, India) and water ad libitum. All the animal experiments were carried out with the prior approval of the Institutional Animal Ethics Committee and were conducted strictly adhering to the guidelines of the Committee for the purpose of Control and Supervision of Experiments on Animals constituted by the Animal Welfare Division of the Government of India.
Cell Line
B16F-10 melanoma, a highly metastatic cell line, was obtained from the National Centre for Cell Sciences, Pune, India. The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) and supplemented with 10% fetal calf serum (FCS) and antibiotics.
Chemicals
DMEM was obtained from Himedia (Mumbai, India). Fetal bovine serum was procured from Life Technologies (Grand Island, NY). Hydroxyproline and glucuronic acid lactone were purchased from Sigma Chemicals (St Louis, MO). N-acetyl neuraminic acid was purchased from Sisco Research Laboratory (Mumbai, India). Oligonucleotide primer sequences were purchased from Sigma Aldrich (Bangalore, India; Table 1). Highly specific quantitative sandwich ELISA kit for mouse interleukin (IL)-1β, IL-2, IL-6, tumor necrosis factor (TNF)-α, and granulocyte macrophage colony-stimulating factor (GM-CSF) were purchased from Pierce Biotechnology (USA), and ELISA kits for vascular endothelial growth factor (VEGF) and tissue inhibitors of MMPs (TIMP-1) were purchased from R & D system (USA). ApopTag Peroxidase in situ apoptosis detection kit was purchased from CHEMICON International Inc (Massachusetts). Cycletest Plus DNA reagent kit was purchased from Becton-Dickson, Canada. All other reagents were of analytical grade.
Oligonucleotide Primer Sequences

Abbreviations: MMP, matrix metalloproteinase; TIMP, tissue inhibitors of MMPs; ERK, extracellular regulated kinases; VEGF, vascular endothelial growth factor.
Preparation of Plant Extract
Authenticated A lanata obtained from Amala Ayurveda Pharmacy was shade dried and powdered. Then, 100 g of powder was subjected to 85% ethanolic extraction in a Soxhlet apparatus for 24 hours. The solvent was evaporated to dryness at 42°C under reduced pressure using a rotary evaporator. The yield of the extract was 16.7% (w/w). 7
The extract was redissolved in a minimum amount of dimethyl sulfoxide for in vitro studies. For animal experiments, it was resuspended in 1% gum acacia and administered intraperitoneally at a dose of 10 mg/kg body weight (based on toxicity studies). The extract was administered using 3 different modalities as follows;
Prophylactic administration: animals were treated with 10 consecutive doses of the drug prior to tumor inoculation.
Simultaneous administration: the drug was given to the animals simultaneously with metastatic tumor cells and continued for 10 consecutive days.
Developed administration: 10 days after tumor inoculation, the drug was administered on 10 consecutive days.
Determination of Antimetastatic Activity in the In Vivo System
Pulmonary colonization assay
C57BL/6 mice were divided into 4 groups (n = 16). Metastasis was induced in all the animals by injecting 106 B16F-10 melanoma cells through the lateral tail vein. To 3 groups of animals, the ethanolic extract of A lanata was administered intraperitoneally at a dose of 10 mg/kg body weight for 10 consecutive days using 3 different modalities: prophylactic administration (group I), simultaneous administration (group II), and developed administration (group III). Group IV animals were untreated metastatic tumor–bearing controls. Eight animals from each group were killed on the 21st day after tumor challenge; the lungs were excised, and blood was collected. The lungs were used for morphological examinations of metastatic tumor nodules and for the estimation of collagen hydroxyproline, 8 hexosamine, 9 and uronic acid 10 contents. Serum was separated from the blood and used for determining the sialic acid 11 and γ-glutamyl transpeptidase (GGT) 12 levels. Serum IL-2, VEGF, and TIMP-1 levels were estimated using the ELISA kit per manufacturer’s protocol. A lung from 1 animal was used for histopathological analysis and from another animal for total RNA isolation. The remaining 6 animals in each group were observed for their survival. The mortality of the animals was observed, and the percentage increase in life span was calculated.
Analysis of the expression of various prometastatic and antimetastatic genes
Total RNA was isolated from the lungs, and cDNA was synthesized using Moloney murine leukemia virus reverse transcriptase. Amplification was performed using specific primers of MMP-2, MMP-9, TIMP-1, TIMP-2, extracellular regulated kinases (ERK)-1, ERK-2, VEGF, and nm23 (Table 1). The amplified products were electrophoresed on a 1.8% agarose gel, stained with ethidium bromide, and photographed under ultraviolet light.
Histopathological analysis
Lung tissues were fixed in 10% formalin, dehydrated in different concentrations of alcohol, and embedded in paraffin wax. Sections (4 µm) were stained with eosin and hematoxylin.
Determination of Antimetastatic Activity Using In Vitro Models
The following parameters were used to assess the antimetastatic action using the in vitro system.
Cell viability
B16F-10 melanoma cells were seeded (5000 cells/well) in 96-well flat-bottomed titer plates and incubated for 24 hours at 37°C in 5% CO2 atmosphere. Different concentrations of A lanata (1-500 µg/mL) were added and incubated further for 48 hours. Then, 4 hours before completion of incubation, 20 µL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (5 mg/mL) was added.13,14 The percentage of viable cells was determined using an ELISA plate reader set to record absorbance at 570 nm.
Proliferation assay
B16F-10 melanoma cells (5000 cells/well) were plated in a 96-well culture plate and incubated at 37°C in 5% CO2 atmosphere. After 24 hours, various concentrations of A lanata (5, 10, and 25 µg/mL) were added and further incubated for 48 hours. 3H-thymidine was added to each well (1 µCi/well) and incubation continued for 18 hours more. After completing incubation, the radioactivity was measured 15 using a Rack Beta liquid scintillation counter.
Collagen matrix invasion assay
The invasion assay was carried out in modified Boyden chambers as described by Albini et al. 16 The lower compartment of the chamber was filled with serum-free DMEM, and a polycarbonate filter coated with 25 µg type I collagen was placed above this. B16F-10 melanoma cells (105 cells/150 µL DMEM) were then seeded on to the upper chamber in the presence and absence of A lanata (5, 10, and 25 µg/mL) and incubated at 37°C in 5% CO2 atmosphere for 10 hours. After incubation, the filters were removed, and the cells were fixed with methanol and stained with crystal violet. Cells migrating to the lower surface of the polycarbonate filters were counted under a microscope. The results were expressed as percentage inhibition of invasion.
Migration assay
Analysis of the effect of A lanata on the migration of the tumor cells was carried out using a Boyden chamber. The polycarbonate filters were placed in Boyden chambers. The lower compartment of the chamber was filled with serum-free DMEM, and B16F-10 cells (105 cells/chamber) suspended in DMEM were then seeded on to the upper chamber in the presence and absence of A lanata (5, 10, and 25 µg/mL) and incubated for 10 hours at 37°C in 5% CO2. The number of cells that migrated to the lower compartment was determined, and results are expressed as percentage inhibition.
Gelatin zymography
SDS-PAGE was performed with 5% gelatin incorporated in the separating gel. 17 B16F-10 melanoma cells of subconfluent cultures were incubated with serum-free medium for 24 hours at 37°C in 5% CO2 atmosphere. The conditioned medium was then collected and subjected to zymographic analysis. Then, 50 µL of sample (equivalent to 100 µg protein) was activated with 5 µL trypsin solution (75 µg/mL) in the presence and absence of A lanata (5, 10, and 25 µg/mL) in 0.1 M Tris–HCl, 10 mM CaCl2 buffer (pH 8.0) and incubated for 1 hour at room temperature. Samples were mixed with an equal volume of 2× sample buffer and loaded on to 11% polyacrylamide gels containing 5% gelatin. Electrophoresis was carried out at 4°C with a constant current of 2 mA/tube until the tracking dye reached the periphery. The gels were then washed with 2% Triton X-100 in 0.1 M Tris–HCl, 10 mM CaCl2 at 37°C for 18 hours followed by staining with GelCode Blue stain reagent for 2 hours. Gels were destained to visualize the clear area against the dark background.
Determination of the Effect of A lanata on the Apoptosis of B16F-10 Cells
The effect of A lanata on the apoptosis of B16F-10 melanoma cells were analyzed by various parameters, such as morphological analysis, DNA fragmentation analysis, TUNEL assay, cell cycle analysis, and expression studies of proapoptotic and antiapoptotic genes by reverse transcription polymerase chain reaction (RT-PCR).
Morphological analysis
B16F-10 melanoma cells (5000 cells/well) suspended in DMEM supplemented with 10% FCS, 100 µg/mL streptomycin and penicillin, and 2 mmol/L glutamine were plated in a 96-well flat-bottomed titer plate and incubated for 24 hours at 37°C in 5% CO2 atmosphere. After 24 hours, different concentrations of A lanata (5, 10, and 25 µg/mL) were added to the cells and incubated further for 48 hours under the same conditions. The cells were then washed with phosphate-buffered saline (PBS; pH 7.4), fixed with 5% formalin, stained using hematoxylin and eosin, and observed under phase contrast microscopy; photographs were also taken. Apoptosis was characterized by examining the morphological changes such as chromatin condensation, nuclear condensation, membrane blebbing, or presence of apoptotic bodies.
DNA fragmentation analysis
One million B16F-10 melanoma cells were treated with different concentrations of A lanata (5, 10, and 25 µg/mL) as described in the previous experiment. After incubation, the cells were treated with 0.1 mL lysis buffer (100 mmol/L Tris–HCl, pH 8.0, containing 0.2% Triton-X 100, and 1 mmol/L ethylene diamine tetraacetic acid [EDTA]) for 10 minutes at −20°C. DNA was extracted using the phenol-chloroform method, precipitated with chilled ethanol, and resuspended in Tris/EDTA buffer (10 mmol/L Tris–HCl, pH 8.0 and 1 mmol/L EDTA). DNA samples were separated by electrophoresis in 1% agarose gels. DNA was stained with ethidium bromide and photographed under UV light.
TUNEL assay
TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay was done to detect apoptosis via DNA fragmentation using ApopTag peroxidase in situ apoptosis detection kit. B16F-10 melanoma cells (5000 cells/well) suspended in DMEM supplemented with 10% FCS, 100 µg/mL streptomycin and penicillin, and 2 mmol/L glutamine were plated in a 96-well flat-bottomed titer plate and incubated for 24 hours at 37°C in 5% CO2 atmosphere. After incubation, 25 µg/mL A lanata was added to the cells, and they were incubated for a further 48 hours under the same conditions. The cells were washed in PBS and stained per manufacturer’s protocol.
Determination of the effect of A lanata on cell cycling of B16F-10 melanoma cells
The cell apoptotic rate was measured by quantification of the percentage of cells with sub-G0/G1 DNA levels, as determined via flow cytometry analysis. Briefly, 1 × 106 logarithmically growing B16F-10 cells suspended in DMEM with 10% FCS were seeded in a culture flask. The cells were treated with A lanata (25 µg/mL) and incubated for 24 hours at 37°C in a CO2 atmosphere. After incubation, the cells were washed with PBS and stained with propidium iodide. DNA content was analyzed by a fluorescence-activated cell sorter (Becton Dickinson, FACS Calibur) using the Cycletest Plus DNA reagent kit per the manufacturer’s protocol.
Determination of the Effect of A lanata on the Expression of p53, caspase-9, caspase-3, caspase-8, bcl-2, bid, bax, p21, p27, and cyclin-D1 using RT-PCR
B16F-10 melanoma cells (2 × 104 cells) suspended in serum-free DMEM (250 µL) were seeded in a 96-well titer plate and incubated for 24 hours at 37°C in 5% CO2 atmosphere. A lanata at a concentration of 25 µg/mL was added, and incubation was continued for another 4 hours. cDNA was synthesized using cell to cDNA kit (Ambion Inc, California, USA). Cells were washed with PBS and heated in cell lysis buffer (provided in the kit) to release the RNA into the solution followed by a heating step to inactivate endogenous RNases. The genomic DNA was further degraded by treating with DNase followed by inactivation of DNase by heating at 70°C. RT was performed at 42°C for 50 minutes using Moloney murine leukemia virus RT (supplied along with the kit). Gene expression analysis was done by PCR. The mouse p53, caspase-9, caspase-3, caspase-8, bcl-2, bid, bax, p21, p27, and cyclin-D1 (Table 1) were amplified against GAPDH standard. The cycling conditions used were as follows: 1 minute at 94°C, 1 minute at 58°C, and 1 minute at 72°C for 40 cycles, followed by a 10-minute extension at 72°C. Amplified samples were subjected to electrophoresis in an agarose gel (1.5%) containing 0.5 µg/mL ethidium bromide and photographed under UV light.
Determination of the Effect of A lanata on Proinflammatory Cytokines and GM-CSF Levels
B16F-10 melanoma cells (5000 cells/well) suspended in DMEM supplemented with 10% FCS, 100 µg/mL streptomycin and penicillin, and 2 mmol/L glutamine were plated in a 96-well flat-bottomed titer plate and incubated for 24 hours at 37°C in 5% CO2 atmosphere. A lanata at a concentration of 25 µg/mL was added to the cells, and they were incubated for a further 48 hours under the same conditions. The supernatant was used for the estimation of cytokines; IL-1β, IL-6, TNF-α, and GM-CSF using specific ELISA kits per manufacturer’s protocol.
Statistical Analysis
Values are expressed as mean ± standard deviation. The statistical analysis was done by 1-way ANOVA followed by Tukey’s multiple comparison using Graphpad InStat software. A P value <.05 was considered to be statistically significant.
Results
Antimetastatic Activity in the In Vivo System
Effect of A lanata on the inhibition of lung metastasis and survival
Metastatic tumor-bearing animals treated with A lanata showed significant reduction in tumor nodule formation (Table 2). Metastatic control animals had massive tumor growth and were assigned an arbitrary number of 250. 18 The 3 different modalities of compound administration were found to be significantly (P < .05) effective. Of these, simultaneous mode of administration of ethanolic extract of A lanata (10 mg/kg body weight, intraperitoneal) produced maximum inhibition of lung nodules (70.53%) followed by prophylactic mode of administration (62.36%) and then administration after tumor development (38.51%).

Abbreviation: ILS, increase in life span.
B16F-10 melanoma cells (1 × 106) were injected into each animal via the lateral tail vein. Aerva lanata was administered intraperitoneally for 10 days. Animals were killed on the 21st day and lung tumor nodules counted. For the survival study, death caused by tumor burden was recorded, and the life span was calculated. Values are mean ± standard deviation.
a, b, c: Means without a common superscript differ (P < .05).
Administration of A lanata significantly increased the life span of metastatic tumor–bearing animals (Table 2). The life span was highly enhanced when A lanata was administered simultaneously (65.3%). Prophylactic administration of A lanata enhanced the life span by 48.2%, whereas administration after tumor development increased the lifespan by 27.1%.
Effect of A lanata on lung and serum biochemical parameters of metastatic tumor–bearing animals
The effect of A lanata on lung collagen hydroxyproline, hexosamine, and uronic acid content is shown in Table 3. Tumor-bearing control animals showed an increased level of lung collagen hydroxyproline (21.25 ± 0.94 µg/mg protein), which was significantly (P < .05) reduced in animals treated with A lanata using prophylactic (10.37 ± 0.59 µg/mg protein), simultaneous (8.41 ± 0.52 µg/mg protein), and developed (16.31 ± 0.94 µg/mg protein) modalities.
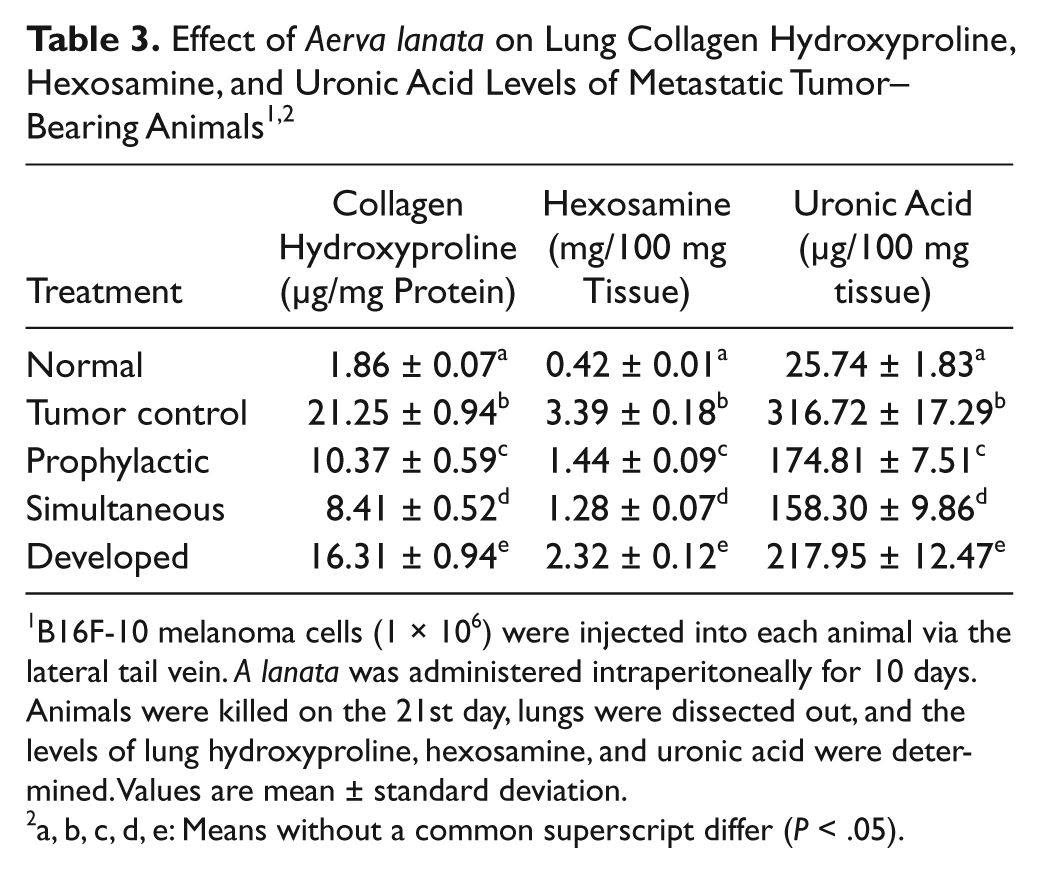
B16F-10 melanoma cells (1 × 106) were injected into each animal via the lateral tail vein. A lanata was administered intraperitoneally for 10 days. Animals were killed on the 21st day, lungs were dissected out, and the levels of lung hydroxyproline, hexosamine, and uronic acid were determined. Values are mean ± standard deviation.
a, b, c, d, e: Means without a common superscript differ (P < .05).
Tumor-bearing control animals had a high level of lung uronic acid content (316.72 ± 17.29 µg/100 mg tissue wet weight; P < .05) when compared with normal animals (25.74 ± 1.83 µg/100 mg tissue wet weight). Treatment of A lanata reduced the uronic acid content in all the 3 modalities: prophylactic (174.81 ± 7.51 µg/100 mg tissue wet weight), simultaneous (158.30 ± 9.86 µg/100 mg tissue wet weight), and developed (217.95 ± 12.47 µg/100 mg tissue wet weight).
Hexosamine content was also high in the lungs of tumor controls (3.39 ± 0.18 mg/100 mg tissue dry weight) when compared with normal animals (0.42 ± 0.01 mg/100 mg tissue dry weight). A lanata treatment significantly (P < .05) reduced the lung hexosamine content in both the prophylactic (1.44 ± 0.09 mg/100 mg tissue dry weight) and simultaneous (1.28 ± 0.07 mg/100 mg tissue dry weight) modality group, whereas it was only 2.32 ± 0.12 mg/100 mg tissue dry weight in the developed-modality group.
The effect of A lanata on serum biochemical parameters is presented in Table 4. The serum sialic acid level of control metastatic tumor-bearing animals was highly increased (134.76 ± 8.39 µg/mL serum) as compared with normal values (22.64 ± 1.03 µg/mL serum). Here also the simultaneous administration of A lanata significantly (P < .05) reduced the elevated serum sialic acid level to 62.86 ± 4.68 µg/mL serum, followed by the prophylactic modality (68.93 ± 3.17 µg/mL serum); in the developed-modality group, it was reduced only to 96.08 ± 5.92 µg/mL serum.

Abbreviations: GT, glutamyl transferase; VEGF, vascular endothelial growth factor; IL, interleukin; TIMP, tissue inhibitor of matrix metalloproteinase.
B16F-10 melanoma cells (1 × 106) were injected into each animal via the lateral tail vein. A lanata was administered intraperitoneally for 10 days. Animals were killed on the 21st day, blood was collected by heart puncture and serum separated, and the levels of serum sialic acid, γGT, IL-2, TIMP-1, and VEGF were determined. Values are mean ± standard deviation.
a, b, c, d: Means without a common superscript differ (P < .05).
Serum γ-GGT level was also drastically (P < .05) enhanced in metastatic control animals (104.27 ± 7.94 U/L) compared with normal animals (3.93 ± 0.24 U/L). After the administration of A lanata, the elevated γ-GGT level was reduced significantly (P < .05) to 63.71 ± 3.29 U/L in the prophylactic group and 58.25 ± 3.65 U/L in the simultaneous-modality group. In animals with developed tumor, the serum γ-GGT level was reduced to 82.64 ± 5.72 U/L by A lanata treatment.
Effect of A lanata on serum IL-2, TIMP-1, and VEGF levels in metastatic tumor–bearing animals
Serum VEGF, IL-2, and TIMP-1 levels are shown in Table 4. The normal serum VEGF level was only 18.37 ± 0.82 pg/mL. This was significantly increased (P < .05) to 163.71 ± 10.2 pg/mL in metastatic tumor–bearing mice. This elevated level of serum VEGF was reduced significantly (P < .05) in the groups with prophylactic (89.04 ± 3.49 pg/mL) and simultaneous (83.53 ± 5.64 pg/mL) modalities. The elevated serum VEGF level was also reduced in animals with developed tumor (107.29 ± 8.61 pg/mL) after A lanata administration. There was a significant reduction in the serum levels of IL-2 (9.84 ± 0.51 pg/mL) and TIMP-1 (384.57 ± 21.43 pg/mL) in tumor-bearing animals. Administration of A lanata using the prophylactic (21.53 ± 1.39 and 583.14 ± 28.62 pg/mL), simultaneous (24.76 ± 1.92 and 603.21 ± 23.79 pg/mL), and developed (27.38 ± 1.06 and 540.86 ± 16.35 pg/mL) modalities significantly (P < .05) restored the levels of IL-2 and TIMP-1 to almost normal levels.
Effect of A lanata on the expression of various prometastatic and antimetastatic genes
Expression of MMP-2, MMP-9, ERK-1, ERK-2, and VEGF genes were downregulated in the groups where A lanata was administered prophylactically and simultaneously with tumor induction (Figure 1). But there was a diminished expression of these genes in animals treated with A lanata after tumor development. The expression of antimetastatic genes TIMP-1 and TIMP-2 was minimal, and nm23 was nearly absent in tumor-bearing animals. Treatment with A lanata resulted in the upregulation in the expression of these genes.

Expression of MMP-2, MMP-9, TIMP-1, TIMP-2, ERK-1, ERK-2, nm23, and VEGF in lungs of metastatic tumor–bearing mice: Lane 1, metastatic tumor control; lane 2, prophylactic Aerva lanata treatment; lane 3, simultaneous A lanata treatment; lane 4, A lanata treatment 10 days after tumor induction
Histopathological Analysis of Lungs
The hematoxylin and eosin stained sections of lung tissues are shown in Figure 2 (×100 magnification). Lungs in the control animals showed infiltration of the neoplastic cells around the main bronchioles extended to the pleura. This together with fibrosis reduces alveolar space, which in turn leads to reduced vital capacity. Prophylactic and simultaneous administration of A lanata resulted in significant reduction in tumor mass. The alveoli and pleura were tumor free, and the alveolar passage was lined with healthy ciliated columnar epithelial cells, almost similar to the normal lung. Considerable reduction in tumor mass was also observed in developed modalities of administration.

Lungs of metastatic tumor-bearing mice: A. lung tumor nodule formation: (a) normal; (b) tumor control with nodules; (c) Aerva lanata, prophylactic treatment; (d) A lanata, simultaneous treatment; (e) A lanata treatment 10 days after tumor induction. B. Histopathology of lung of metastatic tumor-bearing animals (×100 magnification): (a) normal; (b) tumor control with nodules; (c) A lanata, prophylactic treatment; (d) A lanata, simultaneous treatment; (e) A lanata treatment 10 days after tumor induction. C. Survival plot
Determination of Antimetastatic Activity in the In Vitro System
Cell viability assay
Cytotoxicity of A lanata toward B16F-10 melanoma cells in culture is shown in Table 5. At concentrations of 10 and 25 µg/mL, A lanata was found to be nontoxic to B16F-10 melanoma cells, and these concentrations were used for further in vitro experiments.

B16F-10 melanoma cells (5000 cells/well) were plated in a 96-well titer plate with different concentrations of extract and incubated for 48 hours. The percentage of viable cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT) assay. Values are means of triplicate.
a, b, c, d, e: Means without a common superscript differ (P < .05).
Tumor cell proliferation
The effect of A lanata on the proliferation rate of B16F-10 melanoma cells was determined by the 3H-thymidine incorporation assay. Thymidine incorporation is proportional to the potential of the cells to synthesize DNA. The rate of proliferation is expressed as radioactive counts per minute (cpm; Table 6). Untreated B16F-10 cells had very high rates of proliferation (3981.5 ± 73.8 cpm). Administration of A lanata at a concentration of 25 µg/mL significantly (P < .05) reduced the proliferation (3095 ± 49.2 cpm; 22.26%) of B16F-10 melanoma cells. Considerable inhibition in the proliferation of tumor cells was also observed when A lanata was administered at a concentration of 10 µg/mL (3602.5 ± 62.8 cpm; 12.24%).

B16F-10 cells (5000/well) were incubated with different concentrations (25, 10, and 5 µg/mL) of A lanata extract. 3H-thymidine was added to each well (1 µCi/well), and incubation was continued for 18 hours. Cells were lysed, and radioactivity was measured using a Rack Beta liquid scintillation counter. Values are mean ± standard deviation.
a, b, c: Means without a common superscript differ (P < .05).
Collagen matrix invasion assay
Metastatic B16F-10 melanoma cells show a highly invasive property through the collagen matrix. Very high numbers of cells were found in the lower surface of the polycarbonate membrane, but administration of A lanata produced significant inhibition in the invasion of the collagen matrix by the tumor cells in a dose-dependent manner. At a concentration of 25 µg/mL, A lanata significantly inhibited the invasion of B16F-10 melanoma cells by 53.76%, whereas at concentrations of 10 and 5 µg/mL, the percentage inhibition of invasion was found to be 39.49% and 16.18%, respectively (Figure 3).
Migration
Inhibition of tumor cell migration by A lanata is given in Figure 3. A lanata significantly inhibited the migration of B16F-10 melanoma cells across the polycarbonate filters in a dose-dependent manner.
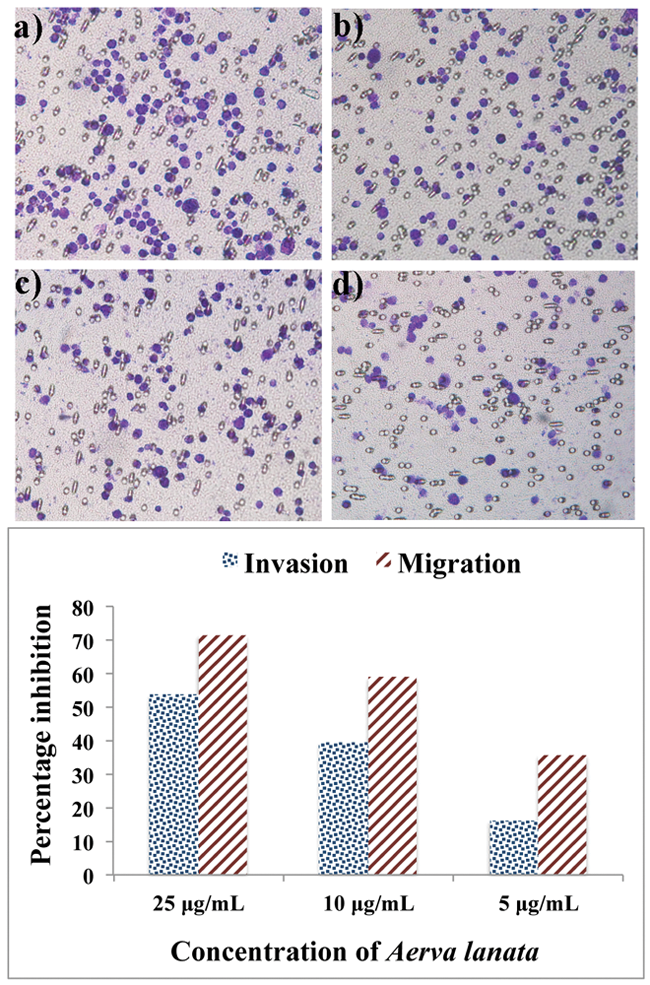
The effect of Aerva lanata on tumor-cell collagen matrix invasion and migration: A. collagen matrix invasion. (A) untreated control; (B) treatment with A Lanata, 5 µg/mL; (C) treatment with A Lanata, 10 µg/mL; (D) treatment with A Lanata, 25 µg/mL. B. Percentage inhibition of tumor-cell invasion and migration by A lanata
Gelatin zymographic analysis
A lanata inhibited the activation of MMPs produced by B16F-10 melanoma cells as shown in Figure 4. Conditioned medium after trypsin activation showed digested clear areas at 92 and 72 kD, which was identical to MMP-9 and MMP-2 activity (Figure 4, Lane 2). Gels loaded with conditioned medium, without trypsin activation, did not show any clear areas, indicating the inactive form of the enzyme collagenase (Figure 4, Lane 1). Trypsin-activated conditioned medium–loaded gels, after incubation with 10 mM EDTA, did not show clear areas, which indicates that the enzyme responsible for degradation is metalloproteinase (Figure 4, Lane 3). When the conditioned medium was treated with A lanata during trypsin activation, no clear bands were observed (Figure 4, Lane 4 and Lane 5 ), indicating that A lanata inhibited the activation of procollagenase to active collagenase at concentrations of 25 and 10 µg/mL. Conditioned medium from cells treated with A lanata at a concentration of 5 µg/mL are shown as 2 faded bands (Figure 4, Lane 6).

Effect of Aerva lanata on MMP-2 and MMP-9 production by B16F-10 melanoma cells: Lane 1. Conditioned medium from untreated B16F-10 melanoma cells without trypsin activation. Lane 2. Conditioned medium from untreated B16F-10 melanoma cells after trypsin activation. Lane 3. Conditioned medium from untreated B16F-10 melanoma cells after trypsin activation + EDTA. Lane 4.Conditioned medium from pretreated B16F-10 melanoma cells (5 µg/mL A lanata) after trypsin activation. Lane 5. Conditioned medium from pretreated B16F- 10 melanoma cells (10 µg/mL A lanata) after trypsin activation. Lane 6. Conditioned medium from pretreated B16F-10 melanoma cells (25 µg/mL A lanata) after trypsin activation
Effect of A lanata on Inducing Apoptosis of B16F-10 Cells
Apoptotic cells were characterized by cell shrinkage, DNA fragmentation, membrane blebbing, and the presence of apoptotic bodies. B16F-10 cells treated with different doses of A lanata (5, 10, and 25 µg/mL) showed the presence of apoptotic bodies and membrane blebbing in a dose-dependent manner (Figure 5). The maximum effect was on the cells treated with 25 µg/mL A lanata. The observed dose-dependent enhancement of DNA fragmentation in A Lanata–treated cells when compared with the untreated controls also supports the above observation (Figure 5).

Effect of Aerva lanata on the apoptosis of B16F-10 cells: A. Morphology of B16F-10 melanoma cells: (a) control, (b) 5 µg/mL A lanata, (c) 10 µg/mL A lanata, (d) 25 µg/mL A lanata. B. DNA ladder formation: lane 1, molecular weight marker; lane 2, control; lane 3, 5 µg/mL A Lanata; lane 4, 10 µg/mL A Lanata; lane 5, 25 µg/mL A lanata. C. TUNEL assay: (a) untreated control after 48 hours of incubation, (b) treated with 25 µg/mL A lanata for 48 hours
TUNEL assay
TUNEL assay has confirmed the presence of apoptotic bodies by staining free 3′-OH termini using enzymatically labeled nucleotides, supporting the above observation (Figure 5). These new DNA ends that are generated on DNA fragmentation are typically localized in morphologically identifiable nuclei and apoptotic bodies. In contrast, the normal B16F-10 cells (controls) that have relatively insignificant numbers of DNA 3′-OH ends did not stain in the above experiment.
Effect of A lanata on Cell Cycle Analysis
Figure 6 shows the effect of A lanata on the B16F-10 cell cycle. In the untreated controls, 3.29% of cells were in the sub-G0/G1 phase, 56.5% of the cells were in the G0/G1-phase (M1), 14.79% of the cells were in the S-phase, and 17.27% of the cells were in the G2/M-phase (M2). Treatment with 25 µg/mL A lanata increased the percentage of cells in the sub-G0/G1 phase to 28.49% after 24 hours of incubation, whereas the percentage of cells in the G0/G1 phase was reduced to 45.28% in the same period.

Effect of Aerva lanata on cell cycling of B16F-10 melanoma cells: (A) untreated control, (B) treated with 25 µg/mL A lanata for 24 hours. M1, G0/G1 (diploid); M2, G2/M (tetraploid); M3, S (synthetic phase); M4, sub-G0/G1 phase. The population of cells in the sub-G0/G1 phase consists of cellular fragments resulting from apoptosis.
Effect of A lanata on the Expression of p53, caspase-9, caspase-3, caspase-8, bcl-2, bid, bax, p21, p27, and cyclin-D1 Using RT-PCR
Agarose gel electrophoresis of the amplified samples showed (Figure 7) that the expression of p53, caspase-9, caspase-3, bax, p21, and p27 were upregulated in A lanata (25 µg/mL) treated cells compared with the nontreated control cells. Caspase-8 did not show any expression in control and A Lanata–treated cells. Bcl-2 and cyclin-D1 genes showed a diminished expression in A lanata–treated B16F-10 cells.

Effect of Aerva lanata on the expression of p53, caspase-9, caspase-3, caspase-8, bcl-2, bid, bax, p21, p27, and cyclin-D1 genes: lane 1, untreated control; lane 2, treated with 25 µg/mL A lanata for 4 hours
Effect of A lanata on GM-CSF and Proinflammatory Cytokine Levels
The effect of A lanata on GM-CSF and proinflammatory cytokine levels is shown in Table 7. A lanata significantly inhibited the production of TNF-α, IL-1β, IL-6, and GM-CSF by B16F-10 melanoma cells in culture.

Abbreviations: TNF, tumor necrosis factor; IL, interleukin; GM-CSF, granulocyte macrophage colony-stimulating factor.
B16F-10 cells (5000 cells) were cultured in the presence of 25 µg/mL A lanata extract for 48 hours. Concentration of cytokines in the culture supernatant was estimated by ELISA. Values are mean ± standard deviation.
a, b: Means without a common superscript differ (P < .05).
Discussion
Metastasis is the process by which cancer cells migrate from a primary tumor to other sites in the body. It is the leading cause of death in cancer patients, and by the time most cancers are diagnosed, metastasis has already occurred, and the presence of multiple metastases makes complete eradication by surgery, radiation, chemotherapy, or biotherapy nearly impossible. Metastasis involves many distinct, successive steps. Tumor cells must escape from the primary tumor site, enter the bloodstream or the lymph system, evade host defense systems, lodge in a spot that is conducive to growth, and establish a colony by recruiting blood vessels for nourishment. 19
Many plant-derived bioactive constituents, including paclitaxel (from Taxus brevifolia), camptothecin (from Camptotheca acuminata), podophyllotoxin (from Podophyllum emodi), and vinblastine (from Catharanthus roseus), have been developed as potential sources of anticancer agents. 20 Recent scientific efforts have focused on the potential roles of extracts of traditional herbs as alternative and complementary medications for cancer treatment.
Administration of the ethanolic extract of A lanata was found to reduce the number of lung metastases caused by the tail vein injection of highly metastatic malignant murine melanoma cells, B16F-10. Inhibition of tumor nodule formation was highest when animals were treated with the A lanata extract simultaneously with tumor administration. This was validated during the histopathological examination of lungs from tumor control and treated animals. There was a significant increase in the life span of tumor-bearing animals treated with the extract when compared with that of the untreated controls. The biochemical parameters in lungs and serum also validated the inhibition of metastasis by A lanata treatment.
TIMPs act as natural inhibitors of MMPs by tightly binding the MMP in a 1:1 stoichiometric ratio and are associated with normal and pathological extracellular matrix turnover. 21 The level of tissue inhibitors of metalloproteinases (TIMP-1) was reduced in the tumor condition, which was brought back to near normal range by treatment with A lanata. The gene nm23 was the first metastasis suppressor gene described and serves as a prototype for this class of genes. In several different tumor systems, lower levels of nm23 mRNA and protein levels have been associated with a more aggressive/metastatic phenotype in vitro. 22 There was an increase in the expression of antimetastatic genes, nm23, and MMP inhibitors (TIMP-1 and TIMP-2) in extract-treated animals along with downregulation of prometastatic genes such as MMP-2, MMP-9, ERK-1, ERK-2, and VEGF.
The majority of MMPs are secreted as zymogens, in which the latency is maintained by interaction of the cysteine residue in the prodomain sequence PRCGVPC with the catalytic zinc ion. Activation of the enzyme in vivo is mediated through the activity of plasmin, cathepsin G, neutrophil elastase, or cellular oxidative changes. In addition to activation of the zymogen, MMP activity is also regulated at the level of transcription and through inhibition of the activated enzyme. The expression of MMPs is induced by a variety of external stimuli such as cytokines and growth factors, including ILs, interferons, EGF, FGF, VEGF, TNF-α, TGF-β, and so on. 4 Zymographic analysis reveals the effect of A lanata on almost complete inhibition of MMP activity at 25 and 10 µg/mL concentration and reduction in MMP-mediated cleavage of gelatin at 5 µg/mL. Effect of A lanata on MMP production and activity is also evident from the inhibition of collagen matrix invasion by B16F-10 cells.
Ethanolic extract of A lanata showed significant cytotoxic activity toward B16F-10 cells and inhibited the proliferation of B16F-10 melanoma cells in a dose-dependent manner.
Apoptosis is an important way to maintain cellular homeostasis between cell division and cell death. A complex balance of proapoptotic and antiapoptotic factors tightly controls the process of apoptosis. Cancer cells can exploit these factors to bypass the normal physiological checkpoints that would trigger defective cells to undergo apoptosis. Apoptosis is characterized by the presence of distinct morphological features and formation of a ladder of DNA fragments. 23 Our results support marked morphological changes indicative of cell apoptosis, including membrane blebbing, chromatin condensation, vacuole formation, nuclear and cytoplasmic fragmentation, and appearance of a DNA ladder in A lanata–treated cells in a dose-dependent manner.
The tumor suppressor gene p53 has been termed the guardian of the genome, given its essential role in surveillance of DNA damage, regulation of the cell cycle, and regulation of apoptosis. The wild-type p53 gene is essential for regulation of cell growth, and it is easy to understand that in a general sense, loss of p53 function may be involved in the early steps of tumor formation through the survival of cells with genetic mutations. Current evidence suggests that p53 functions to detect DNA damage and subsequently arrest cells in the G1 phase of the cell cycle to allow for repair; however, if the damage cannot be repaired, then apoptotic cell death is triggered. 24 There was a significant increase in the percentage of cells in the sub-G0/G1 phase, according to cell cycle analysis by flow cytometry. Caspases are highly selective cystine proteases that control all steps of apoptosis. Caspases can be grouped into 2 types: initiator caspases and effector or executioner caspases. The initiator caspases (eg, caspase 8, caspase 9, and others) act by activating other procaspases, which become effector caspases. Caspase 3, also referred to as apopain, is the major effector caspase. TUNEL assay of A lanata–treated B16F-10 melanoma cells confirms that these cells have free 3′ hydroxyl groups formed by DNA cleavage during apoptosis.
The upregulation of antiapoptotic genes and the downregulation of proapoptotic genes by intervention of natural products are possible mechanisms to induce apoptosis in cancer cells. Alterations in the Bcl-2 family of proteins, a major apoptosis regulatory protein family, often occur in cancers. 25 There was a decrease in the mRNA levels of bcl-2 in the extract-treated cells. A lanata induced cell apoptosis via the intrinsic pathway through the upregulation of antiapoptotic genes (p53, caspase-3, caspase-9, bid, and bax) and cell cycle regulating genes (p21 and p27), which were not expressed in control cells.
In conclusion, this study clearly demonstrates that the ethanol extract of A lanata strongly inhibits lung metastasis of B16F-10 melanoma cells in vivo, which was mediated through induction of apoptosis, via activation of p53-induced caspase-3–mediated proapoptotic signaling and inducing cell cycle arrest at the sub-G0/G1 phase.
Footnotes
Acknowledgements
The authors sincerely thank Dr Ramadasan Kuttan, Research Director, Amala Cancer Research Centre, for his valuable suggestions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.