
Abstract
Background/Rationale:
Accumulating evidence suggests that the use of angiotensin-converting enzyme inhibitor (ACE-I) medication protects against cognitive decline in the elderly patients. We investigated whether ACE-I use was associated with higher plasma levels of amyloid-β (Aβ), possibly indicating improved Aβ clearance from brain to blood.
Methods:
We measured and compared plasma concentrations of Aβ42, Aβ40, and creatinine in cognitively impaired individuals with amnestic mild cognitive impairment, probable Alzheimer’s disease (AD) dementia, and mixed probable AD/vascular dementia.
Results:
Plasma Aβ42 levels and Aβ42/Aβ40 ratios of participants taking ACE-Is (n = 11) significantly exceeded (t = 3.1, df = 19, P = .006; U = 24, P = .029, respectively) those not taking ACE-Is (n = 10).
Conclusions:
This study is the first to show an association between ACE-I use and increased plasma Aβ42 level and Aβ42/Aβ40 ratio in cognitively impaired individuals. Future investigations should assess whether a possible ACE-I-induced increase in plasma Aβ42 indicates improved Aβ42 clearance from brain that contributes to protection from cognitive decline.
Keywords
Introduction
Amyloid-β (Aβ) is a peptide that accumulates in the brains of individuals with Alzheimer’s disease (AD) and may contribute to the neurodegeneration underlying AD symptoms. 1 Two forms of Aβ have been implicated in the pathogenesis of AD—Aβ42, the initial and predominant form composing plaques within brain tissue, and Aβ40, which largely accumulates in cerebral vasculature causing angiopathy. 2 The ability of individuals with AD to clear Aβ40 and Aβ42 from brain is reduced compared to individuals without AD. 3 The disease-modifying potential of improved Aβ clearance from brain is suggested by cerebrospinal fluid (CSF) biomarker studies of cognitively healthy individuals as well as of individuals with AD through which low CSF concentrations of Aβ42 are interpreted as evidence of reduced clearance of Aβ42 from brain to CSF. Sutphen and colleagues found that lower CSF Aβ42 concentration in cognitively healthy middle-aged children of individuals with AD predicted their eventual development of Aβ brain plaques and cognitive decline over an average period of 6 years. 4 Snider et al reported that lower CSF Aβ42 levels in individuals with mild AD predicted more rapid cognitive decline over an average of 3.5 years. 5 Consistent with these findings, studies have also linked lower plasma Aβ levels with a greater cognitive decline among non-demented elderly individuals, 6 a higher rate of incident AD, 7 and a more rapid cognitive decline in individuals with AD. 8,9 A meta-analysis of studies of plasma Aβ as a predictor of dementia and cognitive decline concluded that, despite significant heterogeneity, plasma Aβ42/Aβ40 ratios predict the development of AD and dementia. 10
There is accumulating evidence that the use of antihypertensive medications that inhibit the renin–angiotensin system, including angiotensin-converting enzyme inhibitors (ACE-Is) and angiotensin receptor blockers (ARBs), is associated with a reduced risk of AD 11 and dementia generally. 12 As ACE-Is and ARBs are relatively safe and inexpensive medications that are already approved for other indications, they could be swiftly and widely repurposed for AD prevention and therapy, provided there is convincing evidence for a disease-modifying effect. The present study focused on ACE-Is, which induce vasodilation by decreasing the production of the vasoconstrictor, angiotensin II. Angiotensin-converting enzyme inhibitors are primarily used to treat hypertension but are also indicated for heart failure, acute myocardial infarction, and diabetic nephropathy. 13 An association between ACE-I antihypertensive use and reduced cognitive decline has been observed in samples of nondemented elderly individuals, 14 individuals with mild cognitive impairment (MCI), 15 and individuals with AD. 16 This protective effect has also been observed in nonhypertensive individuals who are taking ACE-Is for other indications. 17 However, not all studies have shown this protective effect 18,19 ; the mechanisms of protection are unclear, and the effect may 14,20,21 or may not 11,22 be restricted to centrally acting ACE-Is that cross the blood–brain barrier, depending on which population is being studied.
This pilot study was an initial, noninvasive attempt to investigate our hypothesis that ACE-Is and ARBs have a protective effect on AD-related cognitive decline by improving Aβ clearance from brain to blood as indicated by higher plasma levels of Aβ. The rationale for this hypothesis is that ACE-Is and ARBs have been shown to preserve and improve cerebral blood flow (CBF), 23,24 and diminished CBF is posited to play a role in reduced clearance of Aβ from brain. 25,26 Due to limited recruitment of individuals taking ARBs, this study was narrowed to assess only the effects of ACE-Is. To our knowledge, there are no reports of clinical studies investigating the effects of ACE-I or ARB medications on Aβ plasma concentrations.
Methods
This was an observational pilot study of 22 individuals enrolled in IRB-approved studies at the University of Maryland Medical Center and the Baltimore VA Medical Center. Participants were recruited from an ongoing dementia health-care services study and from an older adult psychiatry clinic where they were evaluated for memory complaints. Inclusion criteria were: (1) age 60 years or older; (2) participant or legally authorized representative (LAR) willing and able to provide informed consent to participate; (3) participant able to provide verbal or written assent; (4) participant meets diagnostic criteria for probable AD dementia, dementia due to mixed etiology (probable AD and vascular), or MCI; and (5) participant on a stable regimen for 6 months of either taking an ACE-I or ARB or not taking an ACE-I or ARB. The exclusion criterion was that the participant or LAR is unwilling or lacks capacity to provide informed consent to participate. Dementia diagnoses of probable AD dementia and mixed probable AD/vascular dementia were made using National Institute of Neurological and Communicative Disorders and Stroke and AD and Related Disorders Association criteria 27 and by rating participants on the Clinical Dementia Rating Scale (CDR) and the Mini-Mental State Examination (MMSE). 28 Amnestic MCI diagnosis was made by Petersen’s criteria. 29 Cross-sectional data on demographics, cognitive status, medication use, medical history, blood pressure, and plasma biomarkers were obtained.
Blood was drawn by venipuncture to assay levels of Aβ40, Aβ42, and creatinine from platelet-poor plasma. Blood was drawn into BD Vacutainer whole blood/plasma tubes containing sodium citrate (BD, Franklin Lakes, New Jersey). Tubes were placed on ice immediately, centrifuged at 3000g at 4°C for 15 minutes to obtain platelet-poor plasma, and plasma was then aliquoted into polypropylene tubes and stored at −80°C until assay. Aβ40 and Aβ42 concentrations were measured in single batches by enzyme-linked immunosorbent assay (ELISA) kits, the Human β Amyloid (1-40) ELISA Kit, and the high-sensitive Human β Amyloid (1-42) ELISA Kit (Wako Chemicals USA, Inc, Richmond, Virginia), respectively, according to the manufacturer’s instructions. Creatinine was assayed enzymatically in a single batch using the Dimension Vista System ECREA (Siemens Healthcare Diagnostics, Inc, Newark, Delaware), per the manufacturer’s instructions. Data from 11 participants taking ACE-Is for at least the last 6 months [(+)ACE-I group] were compared to participants who had not taken ACE-Is for at least the last 6 months [(−)ACE-I group]. Only 1 individual taking an ARB was recruited; therefore, this participant’s data were not included in the analysis. Normality of numerical data was assessed using the Shapiro-Wilk test. Parametric or nonparametric statistics including 2-tailed Student t, analysis of covariance (ANCOVA), Mann-Whitney U, Pearson r, and Spearman ρ were used as indicated. χ2 Test was used to analyze categorical data. All tests were 2 tailed, and α was set at P = .05. We used SPSS version 12.0 (SPSS, Inc, Chicago, Illinois) for analyses.
Results
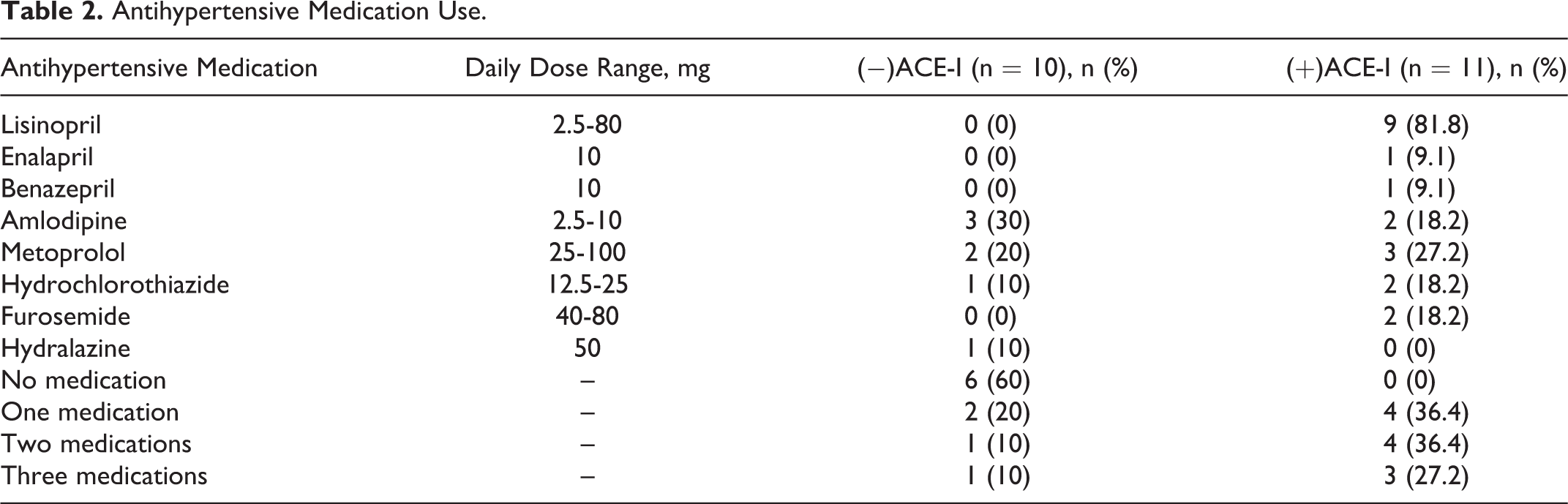
Groups did not differ significantly on demographic variables, CDR sum of boxes scores, MMSE scores, diagnoses, mean plasma creatinine levels, or blood pressure readings (Table 1). In the (+)ACE-I group, lisinopril was by far the most common antihypertensive medication, and 63.6% of the (+)ACE-I group participants were taking more than a single agent (Table 2). Six of 10 (−)ACE-I participants took no antihypertensive medication; however, their blood pressures did not differ significantly from (−)ACE-I participants taking antihypertensives (P > .44) nor from the combined group of (−)ACE-I and (+)ACE-I participants taking antihypertensives (P > .48; data not shown).
Participant Demographics, Clinical Characteristics, and Plasma Levels.
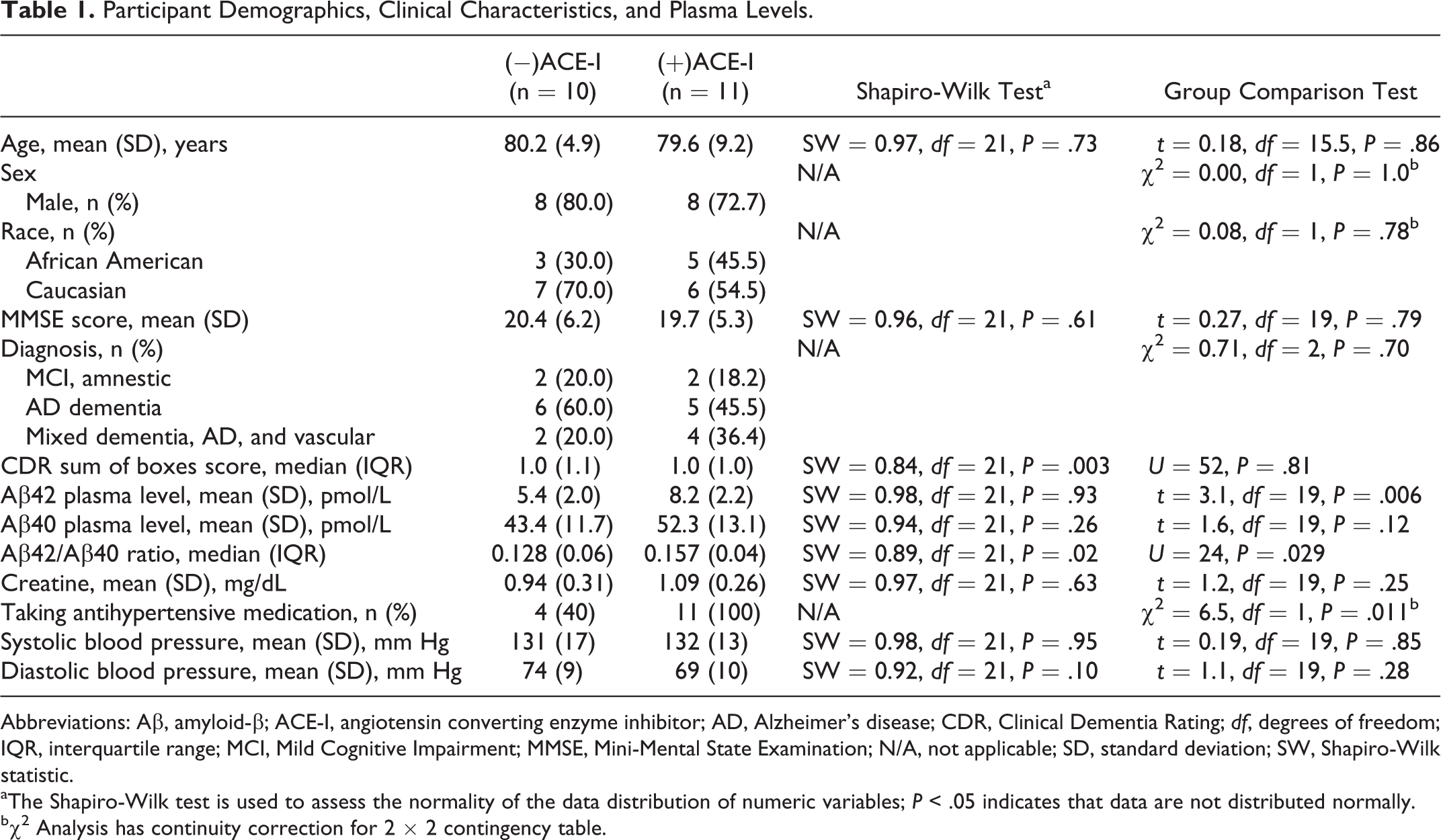
Abbreviations: Aβ, amyloid-β; ACE-I, angiotensin converting enzyme inhibitor; AD, Alzheimer’s disease; CDR, Clinical Dementia Rating; df, degrees of freedom; IQR, interquartile range; MCI, Mild Cognitive Impairment; MMSE, Mini-Mental State Examination; N/A, not applicable; SD, standard deviation; SW, Shapiro-Wilk statistic.
aThe Shapiro-Wilk test is used to assess the normality of the data distribution of numeric variables; P < .05 indicates that data are not distributed normally.
bχ2 Analysis has continuity correction for 2 × 2 contingency table.
Antihypertensive Medication Use.
Average intra-assay coefficients of variation for plasma levels of Aβ42 and Aβ40 were 6.0% and 7.1%, respectively. Consistent with our hypothesis, means for plasma Aβ42 levels and the Aβ42/Aβ40 ratios of the (+)ACE-I group significantly exceeded those of the (−)ACE-I group (Figure 1, Table 1). Analysis of covariance with plasma creatinine level as covariate revealed that creatinine did not predict Aβ42 plasma levels (F = 0.16, P = .700) and the effect of (−)ACE-I versus (+)ACE-I group on Aβ42 levels remained significant (F = 9.3, P = .007). Aβ40 levels of (+)ACE-I participants were not significantly greater than those of (−)ACE-I participants (Figure 2, Table 1). Similarly, ANCOVA with plasma creatinine level as covariate revealed that creatinine did not predict Aβ40 plasma levels (F = 0.53, P = .475) and the effect of (−)ACE-I versus (+)ACE-I group remained insignificant (F = 1.9, P = .189). Plasma Aβ42 and Aβ40 levels correlated positively and significantly (r = .78, P < .001). Aβ42 and Aβ40 plasma levels did not correlate significantly with creatinine plasma levels (r = .08, P = .725; r = .25, P = .282, respectively), lisinopril dose, or any clinical or demographic variable. Average Aβ42 and Aβ40 plasma levels in the 2 participants on the peripheral ACE-Is, enalapril and benazepril (10.3 and 64.7 pmol/L, respectively), were higher than those taking the centrally acting ACE-I, lisinopril (7.8 and 50.6 pmol/L, respectively).
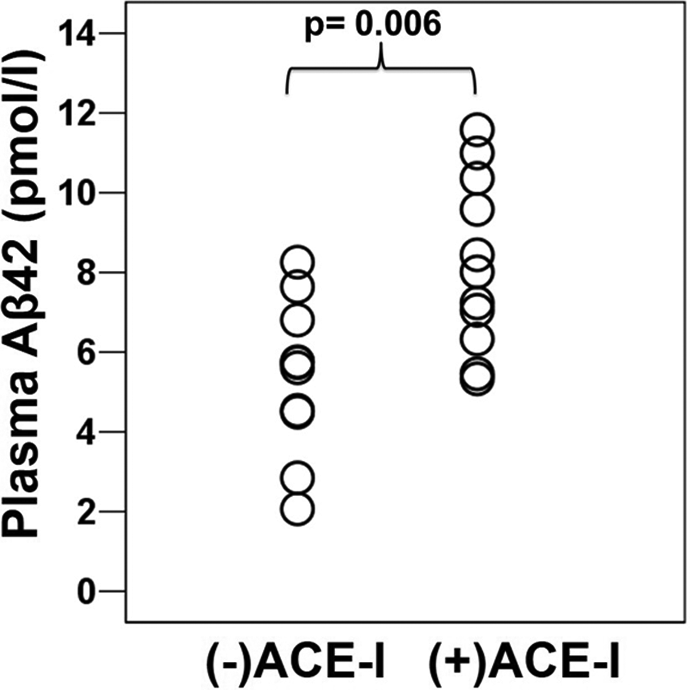
Scatter plot of Aβ42 plasma levels by group.
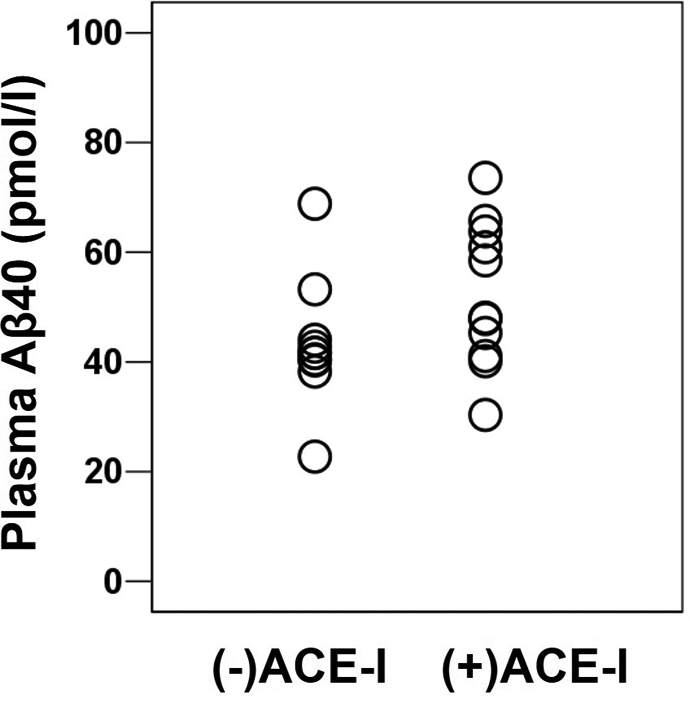
Scatter plot of Aβ40 plasma levels by group.
Discussion
This is the first study to show an association between ACE-I use and increased plasma Aβ42 concentrations and Aβ42/Aβ40 ratios in cognitively impaired individuals. The Aβ42/Aβ40 ratio is thought to be a more sensitive and reliable measure of lower Aβ42 levels, because it normalizes Aβ42 concentration to Aβ40, which as a more plentiful product of amyloid precursor protein (APP) is an approximation of overall Aβ production from APP, thereby enabling detection of low Aβ42 levels in high Aβ producers and vice versa. 30,31 Our results suggest potentially disease-modifying effects of these inexpensive and readily available medications. In addition to using a noninvasive biomarker that could be obtained in routine clinical practice, a number of other study strengths are worthy of note. In particular, groups did not differ statistically on demographic and clinical variables. The similarity between groups in creatinine levels, the lack of correlation between creatinine and Aβ levels, and the ANCOVA results all suggest that disparities in renal function did not contribute to group differences in Aβ plasma levels. However, this study had a number of limitations. The sample size was small, and there were too few participants taking peripheral ACE-Is to meaningfully contrast the effects of peripheral versus centrally acting ACE-Is. Also, most (+)ACE-I participants were not on antihypertensive monotherapy so it is impossible to rule out the effects of other antihypertensives. Finally, there was no central marker of Aβ levels, such as CSF or brain Aβ levels.
Although there are no prior reports of a relationship between ACE-I use and Aβ plasma levels, there is 1 pilot clinical trial that examined the effect of 4 months of the centrally acting ACE-I, ramipril, on CSF Aβ levels in cognitively healthy middle-aged individuals with a parental history of AD. 32 This study found no significant effect of ramipril on CSF Aβ levels despite a significant decrease in CSF ACE activity. The authors noted that the lack of a treatment effect could have been due to several factors including the short duration of the trial, too few participants, inadequate dose, as well as the normal CSF Aβ levels in their sample, indicating an apparent lack of Aβ neuropathology that may have diminished a potential treatment effect in their sample more than in an AD sample.
As the present study was cross-sectional and lacked a central marker of Aβ levels, we are unable to conclude whether the increased plasma Aβ levels in the individuals taking ACE-Is reflect any change in steady state brain Aβ levels that could indicate improved Aβ clearance. Cell line and animal studies have shown differing results on the effects of ACE-Is on brain Aβ homeostasis. There is some evidence that increased Aβ42 plasma levels could indicate increased overall levels of Aβ42 in both brain and blood rather than enhanced Aβ42 clearance from brain. Results from some animal and cell line studies indicate that ACE degrades Aβ and converts Aβ42 to Aβ40 so that ACE inhibition promotes Aβ and Aβ42 accumulation in cells and brain. 33,34 However, in other animal studies, ACE-deficient mice exhibited brain Aβ levels indistinguishable from those of wild-type animals, and treatment of wild-type mice with ACE-Is had no effect on brain or plasma levels of amyloid β. 35 Furthermore, to our knowledge, there is no evidence of increased Aβ brain accumulation with ACE-I use in humans.
In order to put the present study in proper context, it is important to mention that there is extensive animal model evidence of mechanisms other than our hypothesized improved Aβ clearance by which ACE inhibition could exert beneficial effects in AD. For example, the centrally active ACE-I perindopril has been reported to protect against cognitive impairment and brain injury in experimental AD mouse models by suppressing microglia and astrocyte activation and by reducing oxidative stress triggered by brain Aβ deposition. 36 These beneficial effects on cognitive impairment and brain injury as well as the associated reductions in brain inflammation and oxidative stress were not observed for the peripheral ACE-Is enalapril and imidapril. Similarly, administering captopril for 6 months has been found to normalize increased hippocampal ACE activity in AD mice while slowing Aβ plaque accumulation in the hippocampus. 37 The neuroprotective effect of captopril was thought to be mediated by reduced amyloidogenic processing of the APP and decreased hippocampal reactive oxygen species, which are known to drive Aβ generation by activating beta- and gamma-secretases. Finally, there is evidence that ACE inhibition may have beneficial effects in AD by ameliorating neurodegeneration triggered by environmental stress. AbdAlla and colleagues 38 reported that inhibition of ACE by captopril retarded the development of hippocampal tau phosphorylation, signs of neurodegeneration, and APP upregulation, as well as opposing glutamate neurotoxicity by preventing glutamate decarboxylase 2 downregulation in aged rats subjected to 4 weeks of chronic unpredictable mild stress, a model that engenders major pathological features of sporadic AD.
Toward realizing the clinical potential of our findings, work should be done to clarify whether increased plasma Aβ42 reflects improved Aβ42 brain clearance. Amyloid-β detected in plasma can originate from brain as well as from numerous peripheral tissues including blood vessels, skeletal muscle, liver, and platelets. 39,40 Longitudinal measurements of Aβ plasma levels along with brain, and preferably also CSF, Aβ levels are needed to assess the relationship of plasma Aβ levels to central nervous system Aβ levels. Furthermore, clinical trials are required to confirm that ACE-Is do have a protective effect on cognitive decline and to determine whether improved clearance of Aβ42 from brain to blood is involved in this protection. At present, there are mixed results from small clinical trials of ACE-Is in patients with AD that have examined the rate of cognitive decline, and none have assessed plasma or CSF Aβ levels. 41,42 Future studies should consider assessing the relationship of ACE-I therapeutic effects to both Aβ levels and apolipoprotein E (ApoE) genotype. Apolipoprotein E genotype greatly influences risk of sporadic, late-onset AD, with the presence of the ApoE4 allele increasing risk in a dose-dependent manner. 43 Interestingly, the increased AD risk is thought to be mediated at least in part by effects of ApoE4 on Aβ brain clearance. 44,45 Consistent with a relationship between possible ACE-I effects on cognitive decline and Aβ brain clearance, a large retrospective study of 4830 participants found that ACE-I use was associated with a decreased risk of AD only in the absence of an ApoE4 allele. 22
Conclusion
In summary, we found an association between ACE-I use and elevated plasma Aβ42 levels and Aβ42/Aβ40 ratios in individuals with amnestic MCI, probable AD dementia, and mixed probable AD/vascular dementia. This finding is consistent with, but not necessarily indicative of, increased clearance of Aβ42 from brain to blood, which may contribute to a protective effect of ACE-Is on cognition. Further controlled, clinical trials are necessary to determine whether ACE-I medications are therapeutic in AD and whether the therapeutic mechanism involves improved Aβ42 clearance.
Footnotes
Authors’ Note
Previously presented as a poster at the American Association for Geriatric Psychiatry Annual Meeting; March 2016; Washington, DC.
Acknowledgments
The authors thank Dr Richard O’Brien and Ming Li for their expert advice and for their assistance with preliminary Aβ assays.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.