
Abstract
Background:
Long noncoding RNAs have been shown to play crucial roles in cancer biology, while the long noncoding RNA landscapes of pancreatic ductal adenocarcinoma have not been completely characterized. We aimed to determine whether long noncoding RNA could serve as early diagnostic biomarkers for pancreatic ductal adenocarcinoma.
Method:
We conducted a genome-wide microarray analysis on pancreatic ductal adenocarcinoma and their adjacent noncancerous tissues from 8 Chinese patients.
Results:
A total of 3352 significantly differentially expressed long noncoding RNAs were detected. Of total, 1249 long noncoding RNAs were upregulated and 2103 were downregulated (fold change ≥2, P < 0.05, FDR <0.05). These differentially expressed long noncoding RNAs were not evenly distributed among chromosomes in human genome. Hierarchical clustering of these differentially expressed long noncoding RNAs revealed large variabilities in long noncoding RNA expression among individual patient, indicating that certain long noncoding RNAs could play a unique role or be used as a biomarker for specific subtype of pancreatic ductal adenocarcinoma. Gene Ontology enrichment and pathway analysis identified several remarkably dysregulated pathways in pancreatic ductal adenocarcinoma tissue, such as interferon-γ-mediated signaling pathway, mitotic cell cycle and proliferation, extracellular matrix receptor interaction, focal adhesion, and regulation of actin cytoskeleton. The co-expression network analysis detected 393 potential interactions between 80 differentially expressed long noncoding RNAs and 105 messenger RNAs. We experimentally verified 7 most markedly dysregulated long noncoding RNAs from the network.
Conclusion:
Our study provided a genome-wide survey of dysregulated long noncoding RNAs and long noncoding RNA/messenger RNA co-regulation networks in pancreatic ductal adenocarcinoma tissue. These dysregulated long noncoding RNA/messenger RNA networks could be used as biomarkers to provide early diagnosis of pancreatic ductal adenocarcinoma or its subtype, predict prognosis, and evaluate treatment efficacy.
Keywords
Introduction
Pancreatic ductal adenocarcinoma (PDAC), an aggressive and fatal malignancy, is one of the most common malignancies and ranks #4 leading causes of carcinoma-related death. Because it is very difficult to diagnose, especially at their early stage, the morbidity and mortality of PDAC remain high. Most patients have already presented with serious local invasion and/or distant metastasis when PDAC is first diagnosed and thereby missing the optimal timing for curative surgical resection. 1,2 The overall survival rate was less than 5%. 3,4
Long noncoding RNAs (lncRNAs), a subset of noncoding RNA transcripts that are longer than 200 nucleotides in length, 5 are extensively distributed in the genome. 6 They have been suggested to participate in various biological processes, such as epigenetic regulation, chromosome imprinting, cell-cycle control, transcription, translation, splicing, and cell differentiation, 7 -10 and therefore show clinical significance. 11 Dysregulation of lncRNAs contribute to a variety of diseases by altering the expression of lncRNA target genes. 12 -18 For instance, HOTAIR, an oncogenic lncRNA, was associated with the pathogenesis and progression of breast cancer, 19 colorectal cancer, 20 and pancreatic cancer. 21 With the development of transcriptome-sequencing technologies, a considerable amount of novel human lncRNAs have been identified and annotated. However, the molecular mechanisms and biological functions of the vast majority of these annotated lncRNAs remain unknown.
With the development of next-generation sequencing, genome-wide transcriptome profiling has become an effective approach to detect novel lncRNAs in various diseases status. Dysregulation of lncRNAs has been observed in several human cancers, such as osteosarcoma, 22,23 hepatocellular carcinoma, 24 gastric cancer, 25 breast cancer, 26 and endometrial cancer. 27 Recent studies have further revealed that lncRNAs could potentially serve as diagnostic and prognostic biomarkers. For example, a set of 6 lncRNAs has been shown to be independent prognostic factors for glioblastoma after adjust for other clinical variables. 28 Seventeen differentially expressed (DE) lncRNAs, named “SubSigLnc-17”can not only discriminate 2 subtypes of diffused large B-cell lymphoma (DLBCL)—germinal center B-cell-like subtype and activated B-cell-like subtype—but also predict the prognosis of DLBCL. 29 Importantly, lncRNAs have been suggested to participate in pancreatic cancer development and progression by promoting cell growth, migration, invasion, and epithelial–mesenchymal transition. 21,30 In a prospective study, Zhou et al uncovered 7 novel lncRNAs that achieved high performance in distinguishing patients with PDAC from nonmalignant pancreas samples in 3 independent cohorts in the United States. 31 However, the genome-wide profiling of lncRNAs and whether lncRNAs or lncRNA/messenger RNA (mRNA) co-expression network may serve as diagnostic or prognostic biomarkers in Chinese patients with PDAC remain unknown. In the present study, we employed human lncRNA and mRNA arrays to determine the genome-wide transcriptome changes in PDAC tissues from a cohort of Chinese patients. Our results provide an overall review of dysregulated lncRNA and their co-expression networks with dysregulated mRNAs in PDAC tissue. We identified several important and experimentally validated DE lncRNAs. These dysregulated lncRNA/mRNA networks could be used as biomarkers to provide early diagnosis of PDAC or PDAC subtype, predict prognosis, and evaluate treatment efficacy in Chinese patients.
Materials and Methods
Patient Recruitment
Eight PDAC patients who did not receive any chemotherapy or other forms of therapy were recruited from Huashan Hospital, Fudan University. All participants provided written informed consent prior to enrollment. All human patient-related protocols were approved by medical ethics committee of Huashan Hospital affiliated to Fudan University. The PDAC tissue and their adjacent noncancerous tissue were obtained surgically. Totally, 16 samples (2 samples/patient) were immediately frozen down in liquid nitrogen and stored in −80°C freezer. Surgically removed tissues were pathologically confirmed with more than 80% viable tumor cells, and clinical data were obtained retrospectively from electronic clinical records.
RNA Extraction
Total RNAs were extracted from 16 samples above using RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s manual. The quantities and integrity were tested by using NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts) and standard denaturing agarose gel electrophoresis.
Microarray and Data Analyses
We utilized Human 46180K lncRNA arrays manufactured by Agilent Technologies (Santa Clara, California) and Sureprint G3 Human lncRNA Chip (ie, BT1000312) for lncRNA and mRNA microarray analysis. These 2 chips have been reported to represent more than 46 506 lncRNAs and 30 656 mRNAs from NCBI RefSeq, UCSC, RNAdb, and newly annotated lncRNAs in the human genome. Each transcript was represented by up to 5 probes to improve statistical confidence. Differentially expressed genes were defined as fold change ≥2, P < .05, FDR <0.05, in PDAC tissues compared to adjacent noncancerous tissues.
Total RNA (200 ng) from each sample was reversely transcribed into complementary DNA (cDNA) using an RNA Spike In Kit with one color (Agilent Technologies) in the presence of 0.8 mL of random primer mix and 2 mL of Spike mix. These cDNA samples were then cleaned and labeled in accordance with the one color Agilent Gene Expression Analysis protocol using Low Input Quick-Amp Labeling Kit (Agilent Technologies). These labeled cDNA samples were used as probes to hybridize to microarrays for 17 hours at 65°C using an Agilent Gene Expression Hybridization Kit in hybridization chamber gasket slides (Agilent Technologies).
Gene Function Analysis
We used Database for Annotation, Visualization, and Integrated Discovery (http://david.abcc.ncifcrf.gov/) to perform Gene Ontology (GO) analysis. 15 We further applied the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.ad.jp/kegg/) and BioCarta (http://www.biocarta.com) to analyze the potential functions of these target genes in the pathways.16,17. The lower the P value, the more significant the correlation; the recommended P value cutoff is .05.
Long noncoding RNA/mRNA Co-Expression Network
To construct lncRNA/mRNA co-expression network, we calculated the Pearson correlation coefficient and R value to evaluate lncRNA/mRNA correlation. The co-expression network was constructed as follows: (1) data preprocessing: use median value of different transcripts for mRNA but keep the original lncRNA value; (2) data screen: select differential expression of lncRNA and mRNA; (3) calculate the Pearson correlation coefficient and use R value to calculate the correlation coefficient between lncRNAs and mRNAs; and (4) selected genes whose correlation R value was greater than 0.99 and drew correlation network using the cystoscope program.
Real-Time PCR
Real-time polymerase chain reaction (PCR) was performed using the ABI Prism 7300 sequence detection system (Applied Biosystems, Foster City, California). The reaction mixture (20 µL) contained 10 ng cDNA template, sense and antisense primers (200 nM each), and 10 µL 2 × SYBR-Green PCR Mix (TaKaRa, TaKaRa Biomedical Technology, Beijing, China). Real-time PCR was performed in triplicates for each sample, and the specificity of PCR product was tested by dissociation curve. Housekeeping gene β-actin was used as the internal control. 2−△△Ct method was used to quantify the relative expressions of each lncRNA and mRNAs. Data were presented as fold changes of transcripts in PDAC tissue compared to noncancerous tissue. A 2-tailed P value <.05 was considered as statistically significant.
Results
DE lncRNAs and mRNA in PDAC Tissues Compared With Adjacent Noncancerous Tissues
A genome-wide investigation of lncRNA and mRNA expressions in 8 pathologically confirmed PDAC tissues and their adjacent noncancerous tissues was performed using Agilent Human 46180K lncRNA arrays and Sureprint G3 Human lncRNA Chip. Clinical characteristics of these 8 patients are shown in Table 1, and none of these patients had metastasis to other part of the body (M0). A total of 63 431 lncRNAs were detected by microarray and were presented in volcano plot for visualization (Figure 1A) and whiskers plots for quantification (Supplemental Figure 1A). We found that the median number of lncRNA detected were similar between PDAC and noncancerous tissue among individuals (Supplemental Figure 1A). A total of 3352 of these lncRNAs were differentially expressed in PDAC tissues (fold change ≥2, P < .05, and FDR <0.05) compared with their adjacent noncancerous tissues (denoted in colorful dots in Figure 1A). Among these DE lncRNAs, 1249 (37.3%) were significantly upregulated with fold changes ranging from 2 to 54.5. The top 5 unregulated genes are: antisense RNA-1(LEMD1-AS1), SH3 and PX domain-containing protein 2A antisense RNA-1 (SH3PXD2A-AS1), AK130538 (lncRNA located in the intronic region of KMT2Cwhich encodes lysine methyltransferase 2C), CTD-2021H9.2, and homeobox A11 antisense RNA-1 (HOXA11-AS1; Supplemental Table 1). A total of 2103 (62.7%) lncRNAs were downregulated with the fold change ranging from 2 to 21.7. The top 5 annotated decreased lncRNAs were GPR50 antisense RNA-1 (GPR50-AS1), linc00458 (intergenic lncRNA), TCONS_00001278 (LYPLAL is the nearest gene), LYPLAL antisense RNA-1 (LYPLAL1-AS1), and RP11-103C3.1. To our knowledge, none of the top 5 up-/downregulated lncRNAs has been functionally uncharacterized.
Clinical Characteristics of Enrolled Patients (n = 8).a

a Clinical characteristics of patients enrolled in the present study. American Joint Committee on Cancer (AJCC) TNM system: T: whether the primary tumor has grown outside the pancreas and into nearby organs size; N: tumor spreads to regional lymph nodes. M: cancer has metastasized to other organs of the body. n = 8 patients.
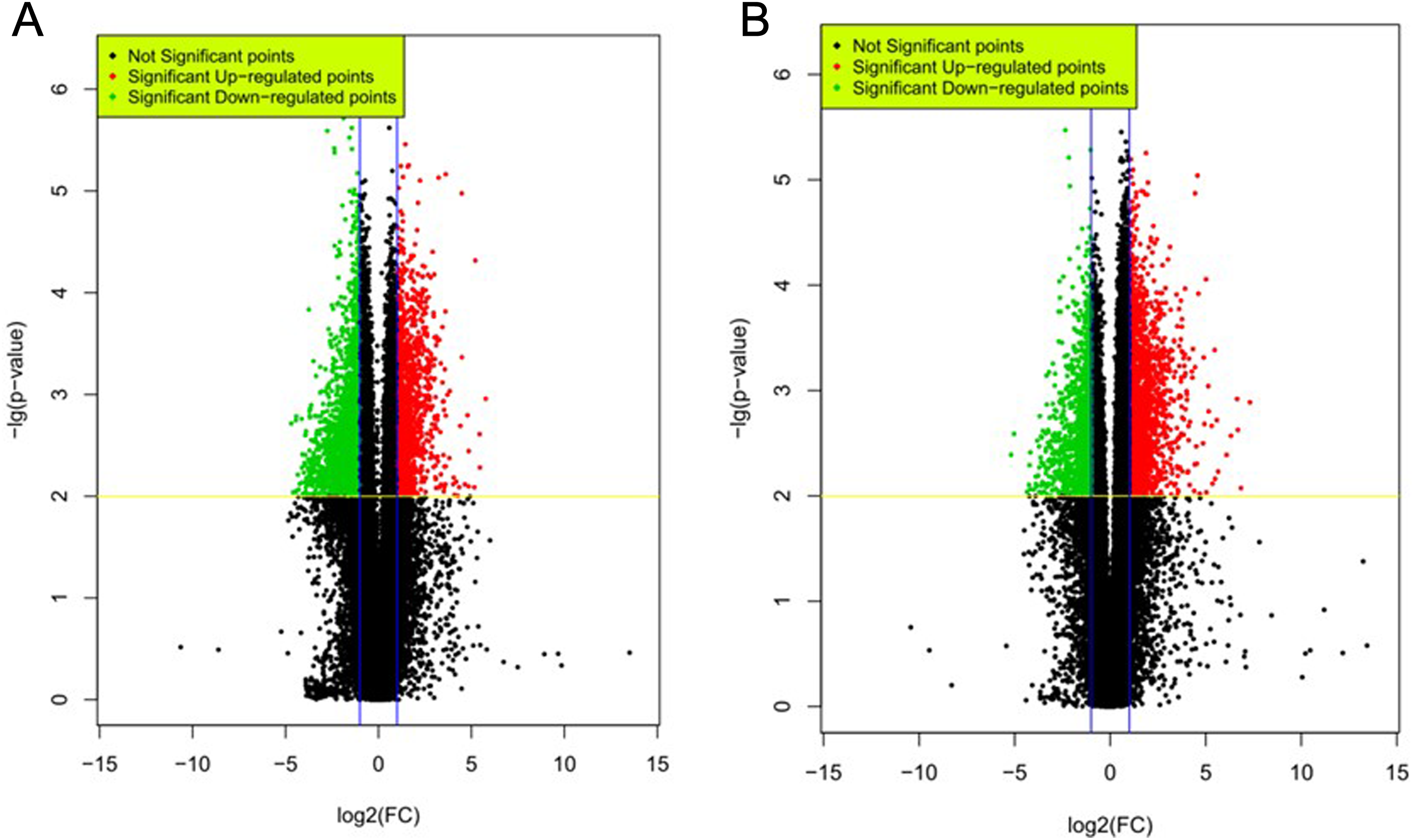
DE lncRNAs and mRNAs in PDAC tissues versus adjacent noncancerous tissue. Shown are the volcano plots of DE lncRNA (A) and mRNA (B). Upregulated and downregulated DE transcripts in PDAC with fold change ≥2, P < .05 and FDR <0.05 are shown in red and green dots, respectively. DE indicates differentially expressed; lncRNAs, long noncoding RNAs; mRNA, messenger RNA; PDAC, pancreatic ductal adenocarcinoma.
In parallel, we analyzed the expressions of coding genes in PDAC versus normal tissues. A total of 39 887 genes were detected by microarray in PDAC tissues, and the median number of mRNA detected was comparable between samples (Supplemental Figure 1B). A total of 2717 mRNAs were significantly changed (fold change ≥2, P < .05, and FDR <0.05). In contrast to lncRNA, the majority of DE mRNA (total 2004, 74%) was upregulated with fold changes ranging from 2 to 114.3 (Supplemental Table 2). Top 5 genes with highest fold changes were claudin-18 (CLDN18), carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5), tripartite motif containing 31 (TRIM31), paired-like homeodomain 1 (PITX1), and melanoma inhibitory activity (Table 2). Seven hundred and thirteen (26%) DE mRNAs were downregulated with fold changes ranging from 2.3 to 36.5. Top 5 genes with were ATPase H+/K+ transporting alpha subunit (ATP4A), glycine N-methyltransferase (GNMT), syntrophin γ 2 (SNTG2), G protein–coupled receptor 50 (GPR50), and aquaporin 8 (AQP8; Table 3).
List of Top 5 Upregulated Coding Genes in PDAC.

Abbreviations: CLDN18, claudin-18; PDAC, pancreatic ductal adenocarcinoma.
List of Top 5 Downregulated Coding Genes in PDAC.

Abbreviation: PDAC, pancreatic ductal adenocarcinoma.
Chromosome Distribution of DE lncRNAs and mRNA
Most lncRNAs are likely to carry out their function in nearby coding genes. We therefore investigated the chromosome distribution of those DE lncRNAs and mRNA in human genome. We found that these DE lncRNAs and mRNA were not evenly distributed among chromosomes (Figure 2A left pie chart and 2B). The proportion of DE lncRNA among all annotated lncRNAs on each chromosome (DE lncRNA density) varies among each chromosome, ranging from 8% on chromosome Y to 25.4% on chromosome 16 (Figure 2C). Differentially expressed lncRNA density does not correlate with DE mRNA density on each chromosome (Pearson correlation P = .2, Supplemental Figure 1). Interestingly, we observed that the number of DE lncRNA and DE lncRNA density was significantly higher in chromosome X than in chromosome Y, pointing to the relative enrichment of dysregulated lncRNA in chromosome X.

Chromosome distribution of DE lncRNA and mRNA in PDAC versus noncancerous tissue. A, Distribution of DE lncRNA and mRNA on each chromosome. B, Number of DE lncRNA or mRNA on each chromosome. C, Number of DE transcripts (lncRNA or mRNA) normalized to total number of annotated transcript (lncRNA or mRNA) on each chromosome. DE indicates differentially expressed; lncRNA, long noncoding RNA; mRNA, messenger RNA; PDAC, pancreatic ductal adenocarcinoma.
Hierarchical Clustering of DE lncRNAs and mRNAs in Pancreatic Adenocarcinoma
To visualize the general expression pattern of DE lncRNA and mRNAs from each patient, we performed hierarchical clustering analysis based on their average expression levels. All DE lncRNAs were grouped into 14 clusters (Figure 3A). Within clusters 1 to 10, DE lncRNAs were generally upregulated in PDAC tissues compared to their adjacent noncancerous tissues except for subject 224 (s224T). This was mainly because these lncRNAs were highly expressed in the noncancerous tissues and did not further increase in PDAC tissue in subject224. Besides, there was a large variability in the expressions of certain lncRNAs among individual PDAC samples. For example, the expression of lnc-MMP3-1 (matrix metalloproteinases (MMPs)) was decreased in PDAC tissue from subject 224, but was increased in PDAC from the other patients (Supplemental Figure 1). Lnc-NFYB-1:1 level was only reduced in PDAC from subject 223 but was increased in the PDAC from the other patients. Differentially expressed lncRNAs in clusters11 to 14 were generally downregulated in PDAC tissues with 2 exceptions, subjects 224 and 225. Although these PDAC samples were pathologically confirmed PDAC cases, the variability of DE lncRNAs among individual PDAC case suggested that these DE lncRNAs could be used as a molecular marker for specific subtype of PDAC.
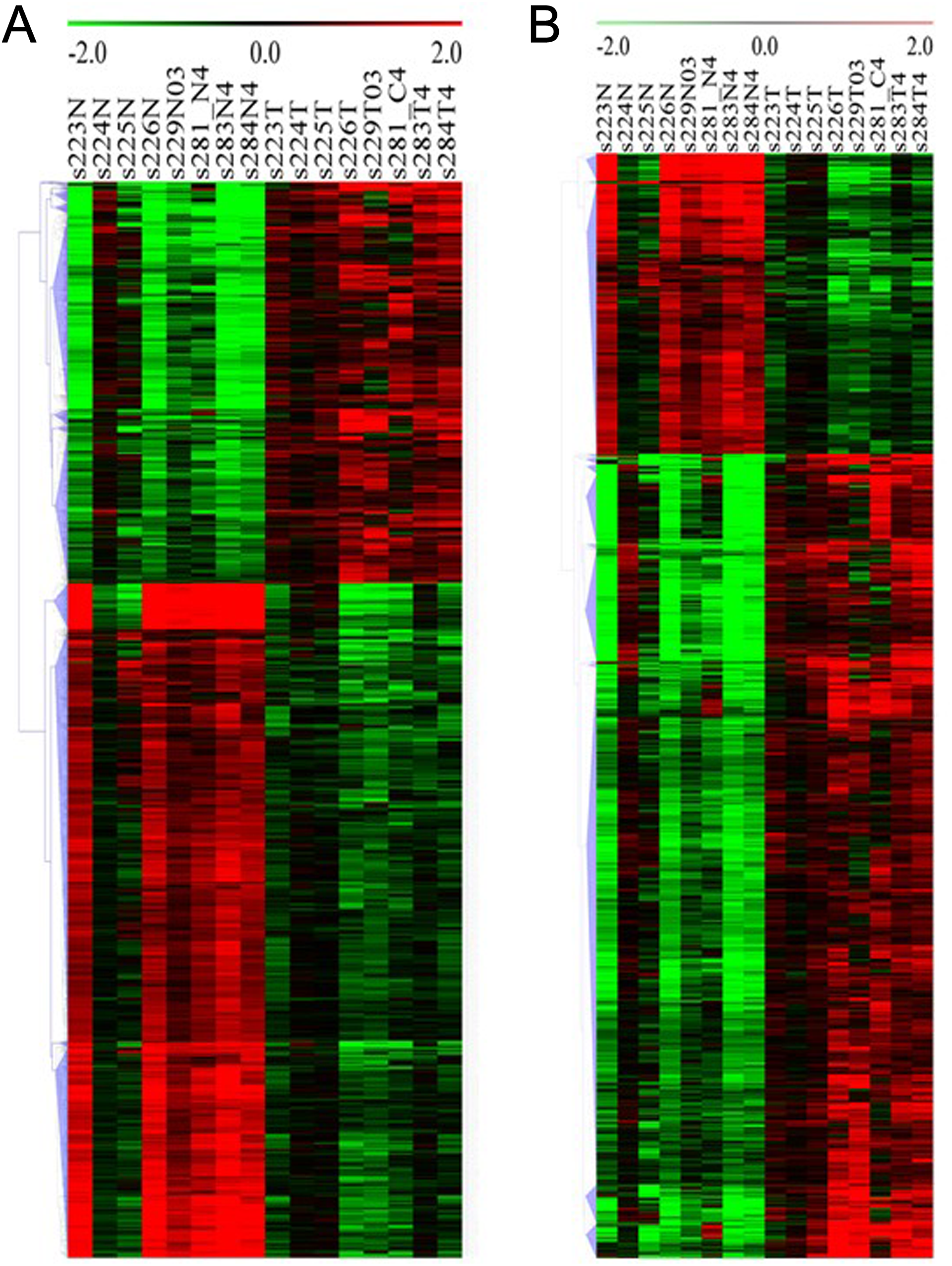
Hierarchical cluster of DE lncRNA (A) and mRNA (B) in human PDAC versus adjacent noncancerous tissue. Clustering was performed based on the average expression levels of transcript from 8 patients. Red: upregulated gene; green: downregulated gene. DE indicates differentially expressed; lncRNA, long noncoding RNA; mRNA, messenger RNA; PDAC, pancreatic ductal adenocarcinoma.
Cluster of DE mRNAs revealed that the majority of DE mRNAs were downregulated, which was opposite to DE lncRNA (Figure 3B; Supplemental Figure 2). Moreover, these mRNAs generally demonstrated no, mild, or moderate difference between PDAC and adjacent cancerous tissue in subject 224 and 225 but displayed more profound difference in the other patients. Variabilities between individuals were also observed. For example, the expressions of HBA2, BTG2, C2orf141, JPH3, and PLCE1 were strikingly higher in PDAC tissue from subject 229 compared to those in the others. The expression of PLIN4, MYRIP, DPT, FGFR1, NTRK2, and NR4A2 was low in almost all PDAC tissues but highly expressed in PDAC from subject 223. These findings further indicated that those genes might play unique and essential role in the pathogenesis of specific subtype of PDAC.
GO Enrichment and Pathway Analysis of DE mRNAs in PDAC
To further explore potential targets of these DE lncRNA in PDAC progression, we performed GO enrichment and pathway analysis on DE mRNAs (Figure 4). Two hundred and eighteen GO terms, including biological processes (127), molecular functions (53), and cellular components (38), were detected and targeted by 9649 nearest coding mRNAs. The target genes and detailed information are represented in Supplemental Table 3. Thirty-one GO terms, including 20 biological processes, 4 molecular functions, and 7 cellular components, were determined to be significant with the threshold lg (P value) > 4.3 (top 25%) and FDR <0.01 (Figure 4A). The most remarkable GO terms were as follows: interferon-γ-mediated signaling pathway, regulation of transcription, mitotic cell cycle, and integral to plasma membrane. The KEGG pathway analysis revealed that 26 of total 271 KEGG pathways analyzed were shown to be significant with the threshold lg (P value) > 3.08 (top 25%) and FDR <0.01 (Figure 4B). Outstanding KEGG pathways include extracellular matrix receptor interaction, focal adhesion, phagosome, regulation of actin cytoskeleton, and several immune responses. These pathways have been reported to be involved in tumor formation and progression. 42

GO terms and KEGG pathways significantly enriched in human PDAC. Shown are significantly enriched GO terms (A) and KEGG pathways (B) of DE mRNAs in human PDAC versus adjacent noncancerous tissue. DE indicates differentially expressed; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; mRNA, messenger RNA; PDAC, pancreatic ductal adenocarcinoma.
Construction of the Co-Expression Network Reveals the Potential Targets (mRNA) of DE lncRNA in PDAC
To further gain insights into the biological functions of lncRNA in the complex biological processes and cellular regulation, DE lncRNA/mRNA co-expression network was constructed to investigate the potential interaction between mRNAs and lncRNAs. As shown in Figure 4A, we detected 393 significant connections (co-expression events) in the network, which was composed of 80 DE lncRNAs and 105 mRNAs (r value >0.99 or <−0.99). Of 80 lncRNAs (nodes) included in the networks, the most outstanding co-expression networks are highlighted by stars with red outlines in Figure 5, such as SH3PXD2A-AS1 (11 edges), lnc-BHLHE23-1 (9 edges), lnc-RP11-204N11.1.1-3:9 (8 edges), and RP11-680F8.3 (9 edges). The most remarkable mRNAs (nodes) in the networks were highlighted by stars with blue outlines in Figure 4, such as ATP10B (10 edges), adenylate cyclase activating polypeptide 1 pituitary receptor type I, 9.6-fold downregulated (ADCYAP1R1; 9), ankyrin repeat domain 30B pseudogene 2, 11.5-fold upregulated (ANKRD30BP2; 9 edges), cytochrome P450 2S1, 10.6-fold upregulated (CYP2S1; 9 edges), GPR133 (9 edges), KLK6 (9 edges), MMP1 (9 edges), and UNC5A (9 edges). Those lncRNAs and mRNAs were highly connected and thus to be considered as the hubs of the network, suggesting their important potential roles of lncRNA in regulating these target genes.

DE lncRNA and mRNA co-expression networks in human PDAC versus noncancerous tissue. Each arrow represents correlation R value greater than 0.99 or smaller than −0.99. Remarkable DE lncRNAs were depicted by stars with red outline, and remarkable DE mRNAs were depicted by stars with blue outline. Yellow stars highlighted DE lncRNAs that were experimentally validated by qRT-PCR. DE indicates differentially expressed; lncRNA, long noncoding RNA; mRNA, messenger RNA; PDAC, pancreatic ductal adenocarcinoma; qRT-PCR, quantitative real-time polymerase chain reaction.
As an example, the most remarkably and reliably dysregulated lncRNA in all 8 patients was SH3PXD2A-AS1. This downregulated lncRNA hub was correlated with 11 edges, 8 of which were also significantly and reliably dysregulated in PDAC tissue, including coding UNC-5 netrin receptor A, 13-fold downregulated (UNC5A), CYP2S1, ANKRD30BP2, ADCYAP1R, lnc-RP11-204N11.1.1-3:9 (10-fold downregulated), DLX2-AS1 (13.6-fold downregulated), lnc00458 (20-fold downregulated), and TCONS_00001278 (19.5-fold downregulated). We thus regarded SH3PXD2A-AS1 as a crucial molecule that involved in pancreatic cancer tumorigenesis through regulating these target genes.
Experimental Validation of DE lncRNAs by qRT-PCR
Finally, we selected7DE lncRNAs that were of highest fold changes and were reliably dysregulated across all 8 patients, including SH3PXD2A-AS1 (highlighted by start), AC006372.4, lnc-BHLHE23-1:1, LYPLAL1-AS1, lnc-RP11-204N11.1.1-3:9, distal-less homeobox 2 antisense RNA-1 (DLX2-AS1), and RP11-680F8.3. We performed experimental validation by reverse transcription quantitative PCR (RT-qPCR; Figure 6). We found that 5 of 7 lncRNAs expression patterns were consistent with microarray data (Figure 6 and yellow stars with red outline in Figure 5). However, changes in LYPLAL1-AS1 and RP11-680F8.3 expressions revealed in qPCR analysis were opposite to microarray analysis (Figure 6 and white stars with red outline in Figure 5).

Validation of important DE lncRNAs by qRT-PCR. n = 8 patients, *P < .05, 2-tailed paired Student t test. DE indicates differentially expressed; lncRNAs, long noncoding RNAs; qRT-PCR, quantitative real-time polymerase chain reaction.
Discussion
The functional significance of lncRNAs has been widely recognized, 42 and recent studies have demonstrated that lncRNAs play important roles in carcinogenesis. 43,44 Dysregulated lncRNAs could be a major cause of oncogenesis or a molecular markers for cancer diagnosis, risk stratification, and monitor of therapeutics. 45,46,21,30 Genome-wide microarray survey of lncRNA may also facilitate our overall understanding of carcinogenesis mechanisms when combined with mRNAs profiling. However, lncRNA landscapes and lncRNA/mRNA co-regulation networks have not been investigated in PDAC from Chinese patients. In the present study, we detected 3352 significantly dysregulated lncRNAs, ∼60% of which were downregulated. In contrast to lncRNAs, >60% DE mRNAs were upregulated in PDAC tissue. Correlation analysis further revealed that the majority of lncRNA and mRNAs were negatively correlated, suggesting that lncRNAs were likely to serve as negative regulators of their targeting coding genes. Gene Ontology and KEGG pathway analysis identified essential pathways that underlie the mechanisms of PDAC tumorigenesis. Dysregulated mRNAs were enriched in pathways such as extracellular matrix receptor interaction, focal adhesion, phagosome, and regulation of actin cytoskeleton (Figure 4), all of which has been reported to be involved in tumor formation and progression. 42
We further found that these DE lncRNAs and mRNAs were unevenly distributed in human genomes. Morbidity of PDAC was indicated to be different in gender, and the incidence of PDAC was more common in women than in men. 47 We showed that the total number or the density of DE transcripts was significantly greater on chromosome X than chromosome Y, suggesting that dysregulated lncRNAs and mRNAs on sex chromosome X could be responsible for increased disease morbidity in women. Hierarchical clustering analysis revealed high variability of dysregulated lncRNAs in each patient. This indicates that the lncRNA molecular signatures may facilitate disease subtype classification, disease risk stratification, and prognosis prediction in pathologically confirmed PDAC cases.
One of the top and most reliably upregulated DE lncRNA in PDAC was SH3PXD2A antisense RNA-1 (SH3PXD2A-AS1; ∼40 fold upregulated). Its opposite strand encodes a protein coding gene named SH3PXD2A. SH3PXD2A mRNA was also significantly and reliably upregulated in all 8 PDAC tissues (fold change = 2.22, P = .007). SH3PXD2A mRNA can be translated into tyrosine kinase substrate 5 (Tks5), a scaffold protein with 5 SH3 domains and 1 PX domain. The Tks5 is a substrate of the tyrosine kinase Src and is required for the organization of podosome and invadopodia formation, thus implicated in invasion of tumor cells. 48 We therefore predicted that the enhanced expression of SH3PXD2A might contribute to pancreatic tumorigenesis by promoting tumor cell invasion. Because the fold changes in SH3PXD2A-AS1 (∼40 folds) were much greater than SH3PXD2A mRNA (∼2 folds), SH3PXD2A-AS1 may serve as a more sensitive biomarker for pancreatic cancer invasiveness.
LncRNAs/mRNAs co-expression network analysis revealed that SH3PXD2A-AS1, the most significantly upregulated lncRNA, is the most remarkable hub with 11 correlated edges. The correlated coding mRNAs included several genes that were implicated in cancer tumorigenesis. For example, UNC5A encoded a protein named UNC-5 netrin receptor A, and it was reported to be a P53-induced tumor suppressor in bladder cancer. 49 ANKRD30BP2, which encoded ankyrin repeat domain 30B pseudogene 2 or CTSP-1, was expressed in 58% of prostate tumors and was capable of eliciting a humoral immune response in about 20% of patients with prostate cancer. 50 The strong expression of CYP2S1, a cytochromes P450 family member, was associated with poor prognosis of colorectal cancer. 51 Moreover, SH3PXD2A-AS1-correlated lncRNAs, including lnc-RP11-204N11.1.1-3:9, DLX2-AS1, and lnc00458, were also significantly dysregulated (Supplemental Table 1) in the microarray analysis. These genes were further experimentally validated using qRT-PCR, suggesting the reliability and reproducibility of our microarray analysis pipeline. Functional studies to test the true causality between lncRNA SH3PXD2A-AS and the abovementioned candidate target genes and to elucidate the molecular actions of these molecules were warranted for a more comprehensive understanding in the mechanisms of human PDAC disease progression.
There are several limitations in the present study: (1) Limited sample size in the present study does not allow us to test the values of DE lncRNAs as the biomarker for early PDAC diagnosis or risk stratification. Further observational or prospective population studies with larger sample size is needed; (2) Future molecular characterization of important DE lncRNAs in PDAC tumorigenesis is warranted.
Conclusion
Our genome-wide survey of dysregulated lncRNAs and lncRNA/mRNA co-expression networks in Chinese PDAC tissue revealed hundreds of dysregulated lncRNA and mRNA. These could be used as biomarkers to guide the diagnosis of subtypes of PDAC, predict prognosis, and evaluate treatment efficacy in patients with PDAC.
Chen Jin and Deliang Fu conceived the study; Sijie Hao, Lie Yao designed the experiments; and Sijie Hao, Lie Yao, Jiaxin Huang, Hang He, Feng Yang, and Yang Di conducted the experiments and analyzed the data.
Footnotes
Abbreviations
Acknowledgment
The authors would like to thank all participating patients.
Declaration of Conflicting Interests
The author(s) declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from Shanghai Municipal Commission of Health and Family Planning, China (Grant No. 20134Y081)
Supplemental Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.