
Abstract
Gastric cancer is one of the most commonly occurring cancers worldwide. Investigation of long noncoding RNAs is of increasing interest, particularly in relation to their contribution to progression and prognosis of gastric cancers; however, insufficient studies been performed investigating the part of long noncoding RNAs play in gastric cancer carcinogenesis. Patterns of dysregulated long noncoding RNA and messenger RNA between mucosa gastric cancer and adjacent normal tissues were identified using long noncoding RNAs microarray analysis. Quantitative real-time polymerase chain reaction was conducted as a means to verify the obtained data. Both Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were subsequently used to investigate the function of dysregulated long noncoding RNAs and messenger RNAs. Cis and trans action was used to predict the possible targets of long noncoding RNAs, and a coexpression network was created to simulate the complex intergenic interactions. Ninety-five dysregulated long noncoding RNAs and 123 messenger RNAs were identified, and quantitative real-time polymerase chain reaction was used to validate 6 filtered long noncoding RNAs. Gene Ontology and KEGG pathway analyses identified several remarkably biological processes and signaling pathways, including spliceosome, RNA transport, and ubiquitin-mediated proteolysis. The transcriptional factors MYC, GABPA, and E2F1 were found to play a central function in the long noncoding RNAs process, as indicated by the coexpression network. This study revealed the dysregulated long noncoding RNA profiles of mucosal gastric cancer. The results shed light on the biological function of long noncoding RNAs in gastric cancer pathogenesis. This provides useful information for exploring potential early screening biomarkers in gastric cancer.
Introduction
Gastric cancer is one of the leading causes of cancer-related death worldwide. 1 In China, gastric cancer is considered a disease of significant morbidity and mortality, as high number of patients are diagnosed with advanced stage and subsequently poor prognosis. Established strategies for treatment are based on radical surgery, but the ending is not satisfactory. 2 Mucosal gastric cancer is defined as a cancer confined to the mucosa of the stomach, without regional lymph node metastases. It achieves a satisfactory long-term prognosis compared to advanced gastric cancer. 3 Therefore, methods to reveal the gastric cancers pathogenesis are attracting increasing attention in cancer-related research.
Analysis of human genome sequencing data has discovered that just 2% of all genes encode for proteins, with the remaining transcripts considered as noncoding RNA (ncRNA) sequences. 4 Long noncoding RNAs (lncRNAs) are a subdivision of ncRNAs with a ≥200 nt sequence length. 5 Long noncoding RNAs have been shown to alter the regulation of gene transcription through chromatin rearrangement by chromosomal looping and through the alteration in binding of various transcription factors (TFs). They are thought to be important in processes linked to tumor development, including both the transcriptional and posttranscriptional regulation of cell differentiation, cell cycle distribution controls, and epigenetic modifications. 6 There has been an accumulation of evidence recently, indicating that there is aberrant expression of lncRNAs in numerous types of cancer, including hepatocellular, lung, glioma, and colorectal cancers. 7 Despite the fact that the expression signatures of lncRNAs have been described in advanced gastric cancer, 8 -11 little research has been carried out investigating lncRNAs expression in mucosal gastric cancer, and the underlying pathways regulating gastric mucosa canceration remain poorly understood. Here, we analyzed the difference in lncRNAs and messenger RNAs (mRNAs) expression patterns between mucosal gastric cancer and adjacent normal tissues.
Materials and Methods
Ethics Statement
This study was approved by the Ethics Committee of Jiangsu University (No. KSYY2018019). All patients provided written informed consent prior to enrollment in the study. Patients were recruited between February 2017 and August 2017 at the Kunshan First People’s Hospital affiliated with Jiangsu University. Each of the patients had a confirmed diagnosis of gastric adenocarcinomas (noncardia gastric cancers) with stage T1aN0M0. No patient received chemotherapy or radiotherapy prior to the operation. Surgical treatment comprised radical distal gastrectomy or total gastrectomy following the recommendations of the Japanese Research Society for gastric cancer. The pathological stages of all patients were evaluated by 3 experienced pathologists for histological confirmation according to the TNM system and the National Comprehensive Cancer Network’s Clinical Practice Guidelines (NCCN Guidelines version 1 2018 gastric cancer).
Tissue Collection and RNA Extraction
Tissues were snap-frozen in liquid nitrogen immediately after resection and stored at −80 °C until use. Total RNA was isolated from frozen samples using a mirVana RNA Isolation Kit (Ambion) following the standard protocols. The concentration and integrity were determined with the NanoDrop ND-2000 (Thermo Scientific) and the Agilent Bioanalyzer 2100 (Agilent Technologies), respectively.
Microarray Expression Profiling of LncRNA and mRNA
OE Bioinformatics Technology Co, Ltd performed all microarray profiling, including labeling of samples, microarray hybridization, and all washing steps, following the standard procedures. Briefly, total RNA was removed leaving pure mRNA after ribosomal RNA was removed by an mRNA-ONLY Eukaryotic mRNA IsolationKit (Epicentre Biotechnologies). Samples were then copied into double-stranded complementary DNA (cDNA), and Cyanine-3-CTP labeled complementary RNA (cRNA) was synthesized. Hybridization of labeled cRNAs onto the Human lncRNA array V6.0 (4×180K, Agilent) was performed, which included the universal profiling of 79 404 lncRNAs and 37 549 coding transcripts. Next, the arrays were washed, and then imaged using an Agilent G2505C Scanner (Agilent Technologies). Array images were assessed, and the raw data were prepared with Feature Extraction software (version 10.7.1.1, Agilent Technologies). Simple analysis of the extracted raw data were performed using Genespring (version 14.8, Agilent Technologies), using the quantile algorithm to normalize the data. Probes identified as having at least one of two parameters highlighted as “P” were selected for additional analysis. Both lncRNAs and mRNAs that were differentially expressed were then recognized through fold-change values, and significance values were determined by Student t test. The limits for upregulated and downregulated genes were set as P ≤.05 and a fold change of ≥2.0. Finally, lncRNAs and mRNAs with distinguishable expression patterns were identified using hierarchical clustering.
Gene Ontology and KEGG Analysis
Gene Ontology and KEGG analysis were applied to determine the roles of these dysregulated lncRNAs and mRNAs. The prediction of lncRNAs function was determined using a method developed for this study. 12 In brief, mRNAs that were coexpressed for each of the lncRNAs that were differentially expressed were determined, and a subsequent functional enrichment analysis was conducted on each group of identified mRNAs that were coexpressed. The functional terms that were enriched were used to predict the functional term for any particular lncRNA. Pearson correlation was used to identify coexpressed mRNAs of lncRNAs by analyzing the correlations that had a P value <.05. The enrichment of functional terms in the coexpressed mRNAs annotation was determined by hypergeometric cumulative distribution function.
Target Gene Prediction
Cis action was used to predict the potential targets of lncRNAs and the cis-regulation areas were determined using the following procedures. Messenger RNAs that were acknowledged as “cis-regulated mRNAs” for each lncRNA when: (i) the mRNA loci were within a range of 100 kb upstream and downstream of the specified lncRNA, and (ii) The lncRNA-mRNA expression Pearson correlation was statistically significant (a correlation coefficient >0.9 and P value of <.05).
Construction of the Coexpression Network
The correlation between the target genes of TFs and lncRNA coexpression coding genes were analyzed using the Pearson correlation method. Hypergeometric distribution analysis was used to identify GO and pathway enrichment coding genes with a high coefficient. The top 200 reliable and predictive associations between differentially expressed lncRNAs and function prediction terms were selected based on Q-value, which underwent further frequency count, and the annotated GO terms were then identified statistically. The graph of the TFs-lncRNAs-mRNAs web was created with Cytoscape 3.01 software (IBS and Agilent). When the crossing of the 2 groups was sufficient, it was predicted that the lncRNAs identified may be involved in pathways controlled by these TFs. Analysis was performed using the ENCODE data on TFs and their regulatory targets.
Real-Time Quantitative Reverse Transcription-PCR
Total RNA was isolated using an RNeasy Mini Kit (217184, Qiagen), according to the manufacturer’s recommendations. The concentration and integrity of the extracted RNA was measured as previously described, and the Superscript III platinum kit (R250-01, Invitrogen) was then used to synthesize cDNA following to manufacturer’s standard instructions. SYBR Green I (CS7561, Invitrogen) was used to carry out real-time polymerase chain reaction (RT-PCR) using an ABIPrism 7500 sequence detector to analyze samples (Applied Biosystems). The following conditions were used as cycle parameters: denaturation at 95 °C for 10 seconds, annealing at 60 °C for 30 seconds, and extension at 70 °C for 45 seconds for 40 cycles. Data were expressed using the comparative threshold cycle method after normalization against β-actin (the house keeping gene). The relative expression of the chosen transcripts was quantified using the 2(−ΔΔCt) method and all experiments were executed in triplicate. Primer sequences were centered on the DNA sequences and designed in the laboratory. The sequences of the primers used were as follows:
XR_250515 F: 5′-GTGTAGATCGAGCCTCCT-3′ XR_250515 R: 5′-GGATGGATTGCTCCAACC-3′ NONHSAT197780 F: 5′-CTGTCATCAAATCTGTCACCAA-3′ NONHSAT197780 R: 5′-TATGCAGTACGAGTTCTGGC-3′ NONHSAT076181 F: 5′-TGGTGTGTGCCTGTAGTC-3′ NONHSAT076181 R: 5′-GGCTCACTGCAACCTCCA-3′ lnc-FAM96A-1 F: 5′-TCTCTTGTAACTCAGGCTGTAG-3′ lnc-FAM96A-1 R: 5′-TTTAGAAACCACCAGCTTGTC-3′ lnc-RAB7A-2 F: 5′-ATCTGGGCACATTGTTGTC-3′ lnc-RAB7A-2 R: 5′-CTGTATCGCTCACCTATCTACT-3′ lnc-OR10H4-3 F: 5′-GCATCTGTCACAATAGACCTAC-3′ lnc-OR10H4-3 R: 5′-TTGTGTGTCTGTGTCAGTTTC-3′ β-actin-F: 5′-CGACATGGAGAAAATCTGGCAC-3′ β-actin-R: 5′-GATAGCACAGCCTGGATAGCAA-3′
Statistical Analysis
Data are all displayed as the mean ± standard deviation. The Student t test was used to analyze the statistical difference between 2 different groups. All statistical tests were executed using SPSS version 17.0. Values of P <.05 were measured as statistically significant.
Results
Dysregulated Expression Profiles of LncRNAs and mRNAs in Mucosal Gastric Cancer
Global transcriptome expression analysis identified 79 404 lncRNAs and 37 549 mRNAs, which included 95 lncRNAs where the expression was significantly changed, with 45 upregulated and 50 downregulated. The list of the top 20 dysregulated lncRNAs was displayed in Table 1. Of the 123 mRNAs found to be dysregulated, 59 were up and 64 were downregulated, respectively. Again, the top 20 dysregulated mRNAs were displayed in Table 2. Hierarchical clustering demonstrated lncRNAs and mRNA expression patterns (Figure 1A). Chromosomal locations of dysregulated lncRNA and mRNAs were shown in Figure 1B.
Top 20 Aberrantly Expressed LncRNA in Microarray for 3 Pairs of Intramucosal Gastric Cancer and Adjacent Noncancer Tissues.

Abbreviations: FC, fold change.
Top 20 Aberrantly Expressed Messenger RNA in Microarray for 3 Pairs of Intramucosal Gastric Cancer and Adjacent Noncancer Tissues.

Abbreviations: FC, fold change.

A, Hierarchical clustering and a heat map of the lncRNA profile comparison between mucosa gastric cancer and adjacent normal tissues. N represents normal tissue and T represents gastric cancer tissue. B, Chromosomal locations of dysregulated lncRNA and messenger RNAs.
Gene Ontology and KEGG Analysis of Dysregulated lncRNAs and mRNAs
The potential biological associations were explored using both GO and KEGG pathway analyses utilizing the 200 lncRNAs that were most dysregulated in mucosal gastric cancer tissues. Gene Ontology investigation suggested that there were numerous enriched functional pathways. Among these pathways, those most closely associated with mucosal cancer were (i) gene expression, (ii) RNA metabolic process, and (iii) spliceosomal small nuclear ribonucleoproteins assembly (Figure 2A). Additionally, KEGG pathway analysis found pathways linked to spliceosome, RNA transport, ubiquitin-mediated proteolysis, and so on (Figure 2B). Gene Ontology analysis of differentially expressed mRNAs were somewhat consistent with the lncRNAs and indicated an involvement in biological processes of sodium ion import across plasma membrane, cellular response to cholesterol, and spinal cord motor neuron differentiation, among others. These mRNA products were mainly found in the organelle membrane, cell periphery, and ciliary membrane. The molecular functions of these mRNAs included potassium:proton and sodium:proton antiporter activity and cholesterol binding (Figure 3A). Conversely, pathway analysis revealed that these mRNAs participated in several signaling pathways, including hedgehog signaling pathway, caffeine metabolism, and basal cell carcinoma, among others (Figure 3B).

Gene Ontology (GO) and KEGG pathway analyses of dysregulated lncRNAs between mucosa gastric cancer and adjacent normal tissues. The top 20 GO terms for the dysregulated lncRNAs in each domain: (A) biological process; (B) cellular component; (C) molecular function. (D) The top 20 KEGG terms for dysregulated lncRNAs in mucosa gastric cancer tissues. The annotation terms are displayed on the vertical axis, and the number of genes annotated to the term is represented on the horizontal axis.
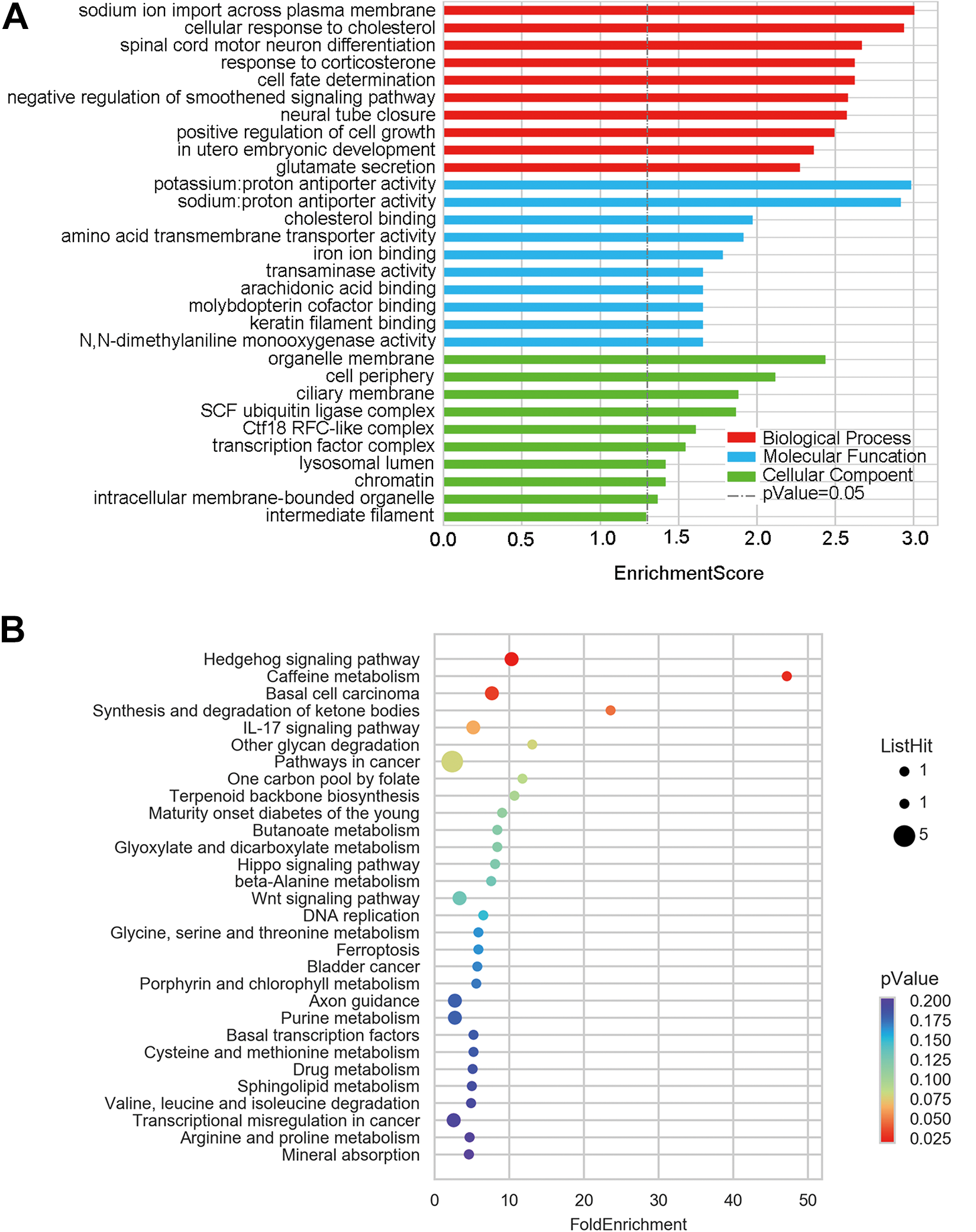
Gene Ontology (GO) and KEGG pathway analyses of dysregulated messenger RNAS (mRNAs) between mucosa gastric cancer and adjacent normal tissues. A, The 10 highest GO terms in each domain for dysregulated mRNAs in mucosa gastric cancer. B, The top 30 KEGG terms for dysregulated mRNAs in mucosa gastric cancer.
Long Noncoding RNAs cis-Regulation
The “cis-regulated mRNAs” of a given lncRNA were defined as the mRNAs whoms loci were within 100 k windows upstream or downstream of a chosen lncRNA, with a Pearson correlation of lncRNA-mRNA expression P value of ≤.05, and 2 lncRNA transcripts corresponding to 2 mRNA transcripts were identified. We concluded that the lncRNA ENST00000497228 may be responsible for the regulation of the expression of upstream coding gene flavin containing monooxygenase 4 (FMO4; Figure 4A). Additionally, the lncRNA ENST00000592710 was established to be able to control the expression of cytochrome P450, family 4, subfamily F, polypeptide 2 (CYP4F2) (Figure 4B).

Fine mapping for the coexpression of lncRNAs and coding genes. The genomic position is identified by an abscissa, a red arrow indicates the genomic position of mRNA transcripts, and a blue arrow represents the location of the lncRNA. An ordinate signifies the correlation coefficient between lncRNA and mRNA transcript. A, Coexpression of ENST00000497228 and coding gene FMO4. B, Co-expression of ENST00000592710 and coding gene CYP4F2. CYP4F2 indicates cytochrome P450, family 4, subfamily F, polypeptide 2.
Coexpression Network Construction
To establish and integral to the gastric cancer pathogenesis, a co-expression network was created centered on the correlation evaluation of mRNAs and lncRNAs that were dysregulated. The network was created using mRNAs and lncRNAs with Pearson correlation coefficients of ≥0.99. Long noncoding RNAs thought to possess trans-regulating functions were analyzed by exploring mRNAs coexpressed with the lncRNAs, with mRNAs that were regulatory targets of specific TFs. The data showed that MYC, GABPA, and E2F1 possibly play important roles in the processes of lncRNAs (Figure 5). The coexpression complex showed the correlation between mRNAs, lncRNAs, and TFs and also revealed the interregulation of lncRNAs and mRNAs in gastric cancer pathogenesis (Figure 6).
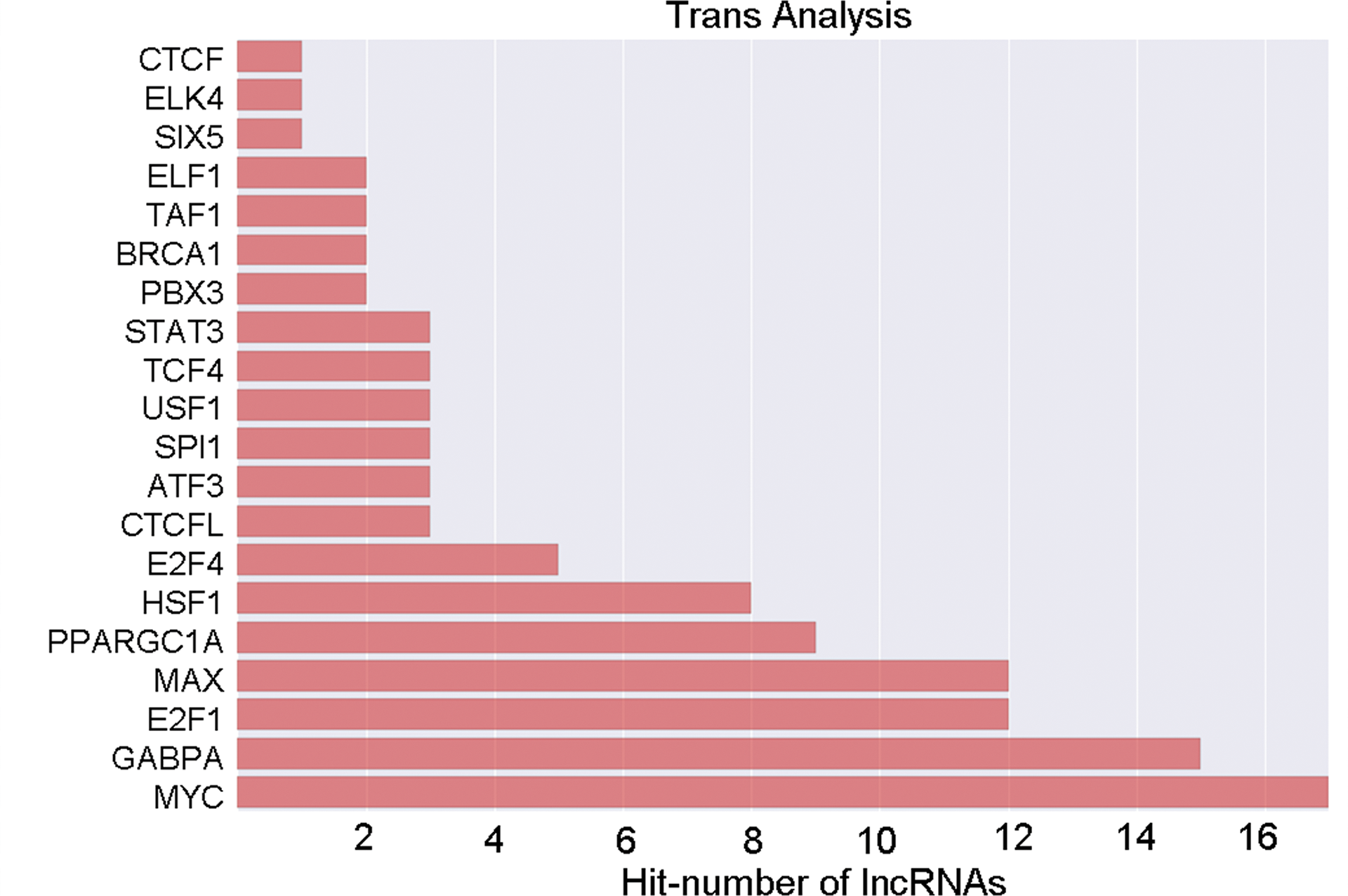
The transcriptional factor profiling of the top results based on aberrantly expressed lncRNAs in mucosa gastric cancer tissues.

Transcription factors (TFs)-lncRNA-mRNA networks. Green, red, and blue nodes represent mRNAs, lncRNAs, and TFs, respectively.
RNA Microarray Validation by Quantitative RT-PCR
To authenticate RNA microarray data, 10 pairs of mucosal gastric cancer and adjacent normal tissues were assessed by quantitative RT-PCR (qRT-PCR) to verify the altered expression of the 6 filtered lncRNAs. The results showed that in mucosal gastric cancer, XR_250515, NONHSAT197780, lnc-FAM96A-1, lnc-RAB7A-2, and lnc-OR10H4-3 were downregulated, while NONHSAT076181 was upregulated. The variation tendency of each lncRNA was consistent with the RNA microarray result (Figure 7).

The validation of 6 dysregulated lncRNAs by qRT-PCR. The relative expression levels of lncRNA in 10 pairs of mucosa gastric cancer and adjacent normal tissues. *P < .05. qRT-PCR indicates quantitative real-time polymerase chain reaction.
Discussion
Gastric cancer is a major health concern, currently being listed as the second leading cause death linked to cancer and the fourth most common cancer worldwide. The progression of gastric cancer is a complex process that involves several genetic and epigenetic modifications. 13 The main reason for the high mortality rate is due to the fact that diagnosis is usually made when the disease is at an advanced stage, as most patients with early-stage gastric cancers are asymptomatic. Typically, the prognosis for patients with advanced gastric cancer receiving the current standard treatment regime remains poor. The development of novel treatment strategies, identification of new molecules for targeted therapy, and the creation of more effective screening techniques to detect this disease at early stage are urgently needed to improve prognosis and lower mortality rates.
High-throughput sequencing has been vital in identified a great number of transcribed lncRNAs throughout the human genome. There is significant accumulating evidence demonstrating that lncRNAs can perform multiple roles in regulating the expression of genes at the transcriptional, posttranscriptional, and even the epigenetic level. 14 The irregular expression of lncRNAs inevitably results in abnormal expression of genes, which could potentially lead to tumorigenesis. Several researchers have identified many different dysregulated lncRNAs in gastric cancer. 15 Aberrantly expressed lncRNAs play critical roles in gastric cancer development and aggressive progression. Currently, there are few studies that have looked into the expression profile of lncRNAs in mucosal gastric cancer, or in establishing the link of lncRNA expression with gastric cancer tumorigenesis.
This study consisted of determining the lncRNAs expression signatures in mucosal gastric cancer samples from patients. A large number of lncRNAs were discovered to be significantly, differentially expressed in gastric cancer, as compared to adjacent normal tissues, utilizing a microarray panel of 79 404 lncRNAs and 37 549 mRNAs, including abundant and varied probes. There were 95 lncRNAs and 123 mRNAs discovered to be significantly differentially expressed. Additionally, the microarray results were validated using qRT-PCR, and findings were consistent, demonstrating the robustness of the data. The results show there are distinctive lncRNAs expression fingerprints in the progression of gastric cancer. In an attempt to gain insight into the functions of lncRNA target genes, both GO and KEGG pathway analyses were carried out on the lncRNA target gene pool. The results demonstrated some pathways, including spliceosome, RNA transport, ubiquitin-mediated proteolysis, potentially play important roles in gastric cancer development. Also, the data revealed that lncRNA ENST00000497228 is associated with FMO4 and ENST00000592710 is linked with CYP4F2, which suggests that lncRNA may exert its functions through regulation of the transcription of related protein-coding genes. Transcription factors-lncRNAs-mRNAs network revealed that MYC, GABPA, and E2F1 performed a vital role in the lncRNAs course and in the occurrence gastric cancer.
For gastric cancer, early diagnosis is the most important way to improve prognosis. Previous a study assessed patients with gastric cancer for the plasma levels of lncRNAs H19, HOTAIR, and MALAT1. 16 Ideally, a biomarker should be accessible noninvasively an identifiable during early stage of the disease. Long noncoding RNAs show potential value as biomarkers in the diagnosis of gastric cancer and in determining the prognosis of the disease. 17 However, there are still many challenges to be faced before lncRNAs can be used in clinical applications. More work is needed to identify further cancer-specific lncRNAs, confirm the usefulness for early diagnosis of lncRNA-based biomarkers, define the roles of specific lncRNAs in gastric cancer tumorigenesis, and to determine if it is possible for lncRNAs to have the potential to improve therapeutic targets.
Conclusions
This study revealed several dysregulated lncRNAs during early stage of gastric cancer. Identification of specific dysregulated lncRNAs show promise as molecular biomarker candidates.
Footnotes
Abbreviations
Authors’ Note
The microarray data used to support the findings of this study are included within the supplementary information file.
Acknowledgments
The authors acknowledge the technical supports provided by OE Bioinformatics Technology Co, Ltd (Shanghai, China).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the youth project “Science and Education” of Suzhou City [KJXW2016062]; the National Natural Science Foundation of China [No.81302386], and Gusu Health Talents Training Fund [GSWS2019083].