
Abstract
This review explores the role of bioinformatics in identifying novel biomarkers for immune cell exhaustion (ICE), a dysfunction state in T cells during chronic infections and cancer. ICE, marked by upregulation of inhibitory receptors such as PD-1 and CTLA-4, impairs immune responses, a critical barrier in chronic infection and cancer treatment. Understanding this state is crucial for developing therapies to reverse T cell exhaustion and improve immune function. The review highlights advanced bioinformatics tools that analyze high-throughput sequencing data, transcriptomics, proteomics, and metabolomics to identify biomarkers and therapeutic targets, enhancing diagnostics and treatments. Despite challenges like the complexity and heterogeneity of ICE, the integration of bioinformatics has advanced our molecular understanding and identification of key pathways. This facilitates the development of personalized immunotherapies, improving outcomes for patients with chronic infections and cancer. Additionally, this review emphasizes the tumor microenvironment’s (TME) role in ICE, where factors such as the upregulation of immune checkpoint ligands, secretion of immunosuppressive cytokines like Transforming Growth Factor Beta (TGF-β) and Interleukin 10 (IL-10), and recruitment of regulatory immune cells create an immunosuppressive milieu fostering tumor growth. In conclusion, this review will also discuss the future directions for research in biomarker discovery and the integration of bioinformatics with clinical data to enhance the precision and effectiveness of therapies. By addressing these challenges, future research can lead to more targeted and efficient treatments for patients suffering from chronic infections and cancer.
Keywords
1. Introduction
Immune cell exhaustion (ICE) is a state of T cell dysfunction that emerges during chronic infections and cancer, characterized by a progressive decline in effector functions and the sustained expression of inhibitory receptors. Immune exhaustion was first recognized in the context of chronic viral infections, such as HIV and hepatitis C, where T cells become persistently exposed to antigens, leading to diminished immune responses. 1 The phenomenon is similarly observed in cancer, where the exhaustion of T cells enables tumors to evade immune surveillance, facilitating their growth and progression. 2 The importance of understanding ICE lies in its implications for disease management and therapeutic development. Global bibliometric analyses of ICI research highlight that immune cell exhaustion and TME modulation are the fastest-growing subfields, while immune-related adverse events (IRAEs) remain a critical barrier to clinical success—underscoring the need for biomarkers that predict both efficacy and toxicity.3,4
ICE manifests through the upregulation of multiple inhibitory receptors, such as P Programmed Death-1 (PD-1), Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA-4), Lymphocyte Activation Gene-3 (LAG-3), and T-Cell Immunoglobulin and Mucin-Domain Containing-3 (TIM-3). This state also involves significant transcriptional and epigenetic reprogramming of T cells, which further entrenches their dysfunctional status. 5 Recent studies have leveraged advanced bioinformatics tools to identify novel biomarkers and therapeutic targets related to ICE, significantly advancing our understanding and ability to intervene in these processes. 6 Specifically, bioinformatics enables the systematic analysis of high-throughput data to: decode transcriptional or epigenetic signatures of exhausted immune cells; construct regulatory networks linking TME factors to ICE; validate candidate biomarkers via machine learning-driven prediction models, and stratify patients for immunotherapy response. This review focuses on these core applications, highlighting how bioinformatics bridges molecular profiling and clinical utility in ICE biomarker discovery. Bioinformatics has revolutionized the analysis of complex biological data, enabling researchers to decode the molecular underpinnings of ICE more effectively. By integrating high-throughput sequencing data, transcriptomics, proteomics, and metabolomics, bioinformatics approaches can identify molecular signatures associated with ICE that are critical for developing diagnostic, prognostic, and therapeutic tools. 7 Unlike existing reviews that focus on either ICE mechanisms or bioinformatics tools in isolation, this work provides a novel integrative framework: we systematically link bioinformatics methodologies to ICE-specific biomarker discovery and clinical translation, with a dedicated focus on resolving TME-ICE crosstalk. Additionally, we address unmet challenges in multi-omics data integration and heterogeneity, offering actionable strategies to bridge bench-to-bedside gaps—perspectives that remain underemphasized in current literature.
Current challenges in the field include the complexity and heterogeneity of ICE across different diseases and patient populations, which complicates the identification of universal biomarkers. 8 This review also aims to highlight the significance of ICE in cancer immunotherapy, where reversing exhaustion can enhance the efficacy of treatments such as checkpoint inhibitors and adoptive cell therapies. Addressing the challenges and complexities associated with ICE will be pivotal in enhancing the efficacy of cancer immunotherapies and improving patient outcomes.
2. Overview of Immune Cell Exhaustion
2.1. Mechanisms of Immune Cell Exhaustion
ICE is characterized by the progressive loss of T cell effector functions, sustained expression of inhibitory receptors, and altered transcriptional profiles. Exhausted T cells (TEX) commonly express high levels of inhibitory receptors such as PD-1, CTLA-4, LAG-3, and TIM-3, which interfere with T cell receptor signaling and cellular metabolism.9,10 These receptors are crucial in maintaining immune homeostasis, but their chronic engagement leads to T cell dysfunction and the establishment of an exhausted phenotype. The expression of these inhibitory receptors is a hallmark of TEX and a critical factor in their impaired functionality. 11
Recent studies have highlighted the role of the transcription factor Thymocyte Selection-Associated High Mobility Group Box (TOX) in driving the epigenetic and transcriptional changes associated with T cell exhaustion. TOX promotes the expression of multiple inhibitory receptors and supports the establishment of the exhausted state in T cells.
12
Additionally, immune checkpoints such as PD-1 and CTLA-4 play significant roles in TEX by mediating the suppressive signals that contribute to their dysfunction.
13
Furthermore, recent research has identified additional regulators, such as Nuclear Receptor Subfamily 4 Group A (NR4A) and Basic Leucine Zipper Transcription Factor (BATF), which collaborate with TOX to intensify the exhaustion process. NR4A transcription factors are known to limit CAR T cell function in solid tumors, highlighting their role in T cell exhaustion.
14
BATF, on the other hand, has been shown to cooperate with Interferon Regulatory Factor 4 (IRF4) to counter T cell exhaustion in mouse tumor models.
15
These findings underscore the multifactorial nature of transcriptional regulation in ICE and suggest potential therapeutic targets for modulating T cell exhaustion. Understanding the molecular mechanisms underlying T cell exhaustion is essential for developing strategies to reinvigorate exhausted T cells and enhance their anti-tumor and anti-viral functions (Figure 1). Three main pathways are shown to contribute to immune exhaustion. (1). Transcriptional Factor: This pathway includes the transcription factor TOX, which is linked to the inhibitory receptors PD-1, LAG3, CTLA-4, and TIM-3. TOX is depicted as a key factor in the regulation of these receptors. (2). Inhibitory Recepto: This pathway highlights the role of inhibitory receptors such as PD-1, LAG3, CTLA-4, and TIM-3. These receptors are known to be upregulated in exhausted T cells and contribute to the suppression of T cell activity. (3). Epigenetic Change: This pathway involves changes in epigenetics, specifically DNA methylation and histone modification. These epigenetic modifications can alter gene expression and contribute to the establishment and maintenance of the exhausted phenotype.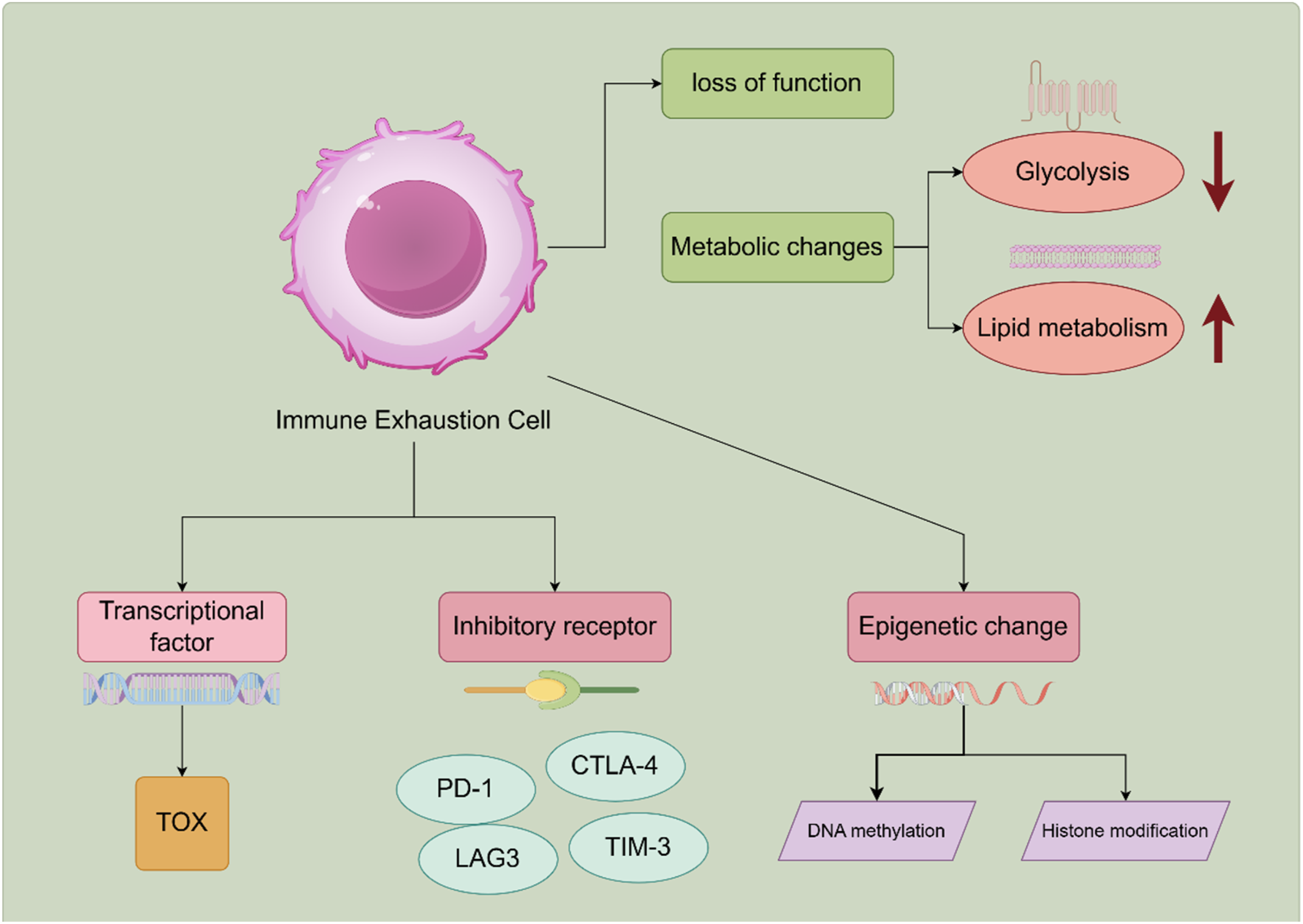
2.2. Clinical Significance of Immune Cell Exhaustion
ICE has significant clinical implications, particularly in the context of chronic viral infections and cancer. In chronic infections like HIV and hepatitis C, exhausted T cells fail to control viral replication effectively, contributing to persistent infection and disease progression. 10 In the tumor microenvironment, T cell exhaustion is a major barrier to effective anti-tumor immunity. Exhausted T cells are often found in high numbers within tumors, where they are unable to exert their cytotoxic functions, allowing tumors to evade immune surveillance and grow unchecked.11,16
The clinical significance of ICE is further underscored by the success of immune checkpoint inhibitors (ICIs) in cancer therapy. ICIs targeting PD-1 and CTLA-4 have shown remarkable efficacy in reinvigorating exhausted T cells and restoring their anti-tumor activity.17,18 However, not all patients respond to these therapies, and understanding the factors that influence the therapeutic outcomes is crucial for improving treatment strategies. For example, the presence of progenitor-exhausted T cells, which retain some proliferative capacity and can be reinvigorated by ICIs, is associated with better clinical responses. 10 Thus, identifying and targeting specific subsets of exhausted T cells and their regulatory mechanisms is vital for enhancing the efficacy of immunotherapies.
3. Bioinformatics Tools and Techniques
3.1. Single-Cell RNA Sequencing (scRNA-Seq)
Single-cell RNA sequencing (scRNA-seq) is a cutting-edge technology that enables the detailed analysis of gene expression at the individual cell level. This method allows researchers to investigate cellular heterogeneity and identify distinct cell populations within complex tissues, such as tumors or inflamed tissues. The basic principles of scRNA-seq involve isolating single cells, converting their RNA into complementary DNA (cDNA), and then sequencing the cDNA to quantify gene expression. This process provides a comprehensive view of the transcriptomic landscape of each cell, facilitating the identification of rare cell types and states. 19
In the context of ICE, scRNA-seq has been instrumental in identifying exhausted T cell subsets and understanding their molecular characteristics. For instance, studies have used scRNA-seq to reveal the heterogeneity among CD8+ T cells in cancer, identifying key exhaustion-associated genes such as PDCD1, TOX, and LAG-3. These findings have provided insights into the functional states of T cells and their responses to immunotherapy. 20 Additionally, advanced scRNA-seq methods, such as multi-omics approaches, combine RNA sequencing with protein profiling to offer a more holistic view of cellular states and interactions. 21 These techniques have significantly advanced our understanding of ICE and paved the way for developing targeted therapies to rejuvenate exhausted T cells. In ICE research, scRNA-seq’s specific utility lies in resolving the heterogeneity of exhausted T cells—for example, distinguishing progenitor exhausted T cells from terminally exhausted subsets in melanoma and NSCLC. This technology has also enabled the discovery of context-specific exhaustion signatures, such as RGS1 as a marker of T cell exhaustion in multiple cancers. However, scRNA-seq has key limitations: its high cost restricts application in large clinical cohorts, low capture efficiency may miss rare exhausted subsets, and batch effects can obscure biological signals without computational correction. 21 Additionally, scRNA-seq alone lacks spatial information, which is critical for understanding how TME localization influences exhaustion—this gap is addressed by integrating scRNA-seq with spatial transcriptomics (Section 3.5).
3.2. Machine Learning and Artificial Intelligence
Machine learning and artificial intelligence (AI) have become powerful tools in the field of bioinformatics, particularly for biomarker identification and the analysis of complex biological data. These technologies can handle large datasets and uncover patterns that may not be apparent through traditional statistical methods. In the context of ICE, machine learning algorithms can be used to analyze scRNA-seq data, identify key regulatory genes, and predict cellular states based on gene expression profiles. 22
Deep learning, a subset of AI, has shown great potential in enhancing our understanding of ICE. By leveraging neural networks, deep learning models can integrate diverse data types, including genomic, transcriptomic, and proteomic data, to predict outcomes and identify novel biomarkers. For example, AI models have been developed to predict patient responses to immune checkpoint inhibitors by analyzing the expression patterns of exhaustion markers and other related genes. 23 These models can help stratify patients based on their likelihood of responding to therapy, thereby improving treatment strategies and outcomes.
Here are some examples:
AI in Single-Cell RNA Sequencing (scRNA-seq): (a) Data AnalysisData Augmentation and Robustness: Generative Adversarial Networks (GANs) are used to generate synthetic scRNA-seq data that resemble real datasets. This helps in augmenting small datasets, improving the robustness and generalization ability of predictive models; (b) Feature Extraction and Clustering: Autoencoders and Graph Neural Networks (GNNs) are employed for efficient feature extraction and clustering of single-cell data. GNNs leverage the intrinsic graph structure of cell relationships to enhance clustering accuracy; (c) Cell Type Annotation: Tools like SingleR use machine learning to automatically assign cell types based on reference datasets. This automates the time-consuming process of manual cell type annotation, making it faster and more accurate.
AI in Network Analysis: Graph Convolutional Neural Networks (GCNs): GCNs are used to analyze the topological structure of networks in ICE research. For example, they can be applied to study the connectivity and dynamics of sea ice networks or cellular interaction networks.
Beyond conventional machine learning, advanced AI models including transformers and graph neural networks (GNNs) have emerged as powerful tools for mining context-specific biomarkers from high-dimensional single-cell data, addressing limitations in capturing complex molecular interactions.
Transformers leverage multi-head self-attention mechanisms to identify long-range gene co-expression patterns and cell-type-specific signatures that are obscured in traditional analyses. For instance, the Transformer-based tool TOSICA enables batch-effect-resistant annotation of million-scale scRNA-seq data, and has been applied to pan-cancer T cell datasets to resolve heterogeneity in exhausted T cell (TEX) subsets—accurately distinguishing progenitor TEX from terminally TEX by prioritizing functional gene modules over individual gene expression. 24 This approach not only identifies core exhaustion biomarkers but also quantifies their regulatory contributions, enhancing biological interpretability.
GNNs, by modeling single-cell data as gene-cell interaction graphs, facilitate context-aware biomarker validation by integrating TME spatial relationships. CellPhenoX, an explainable GNN tool, uses SHAP values to quantify cell subset contributions to clinical outcomes; in triple-negative breast cancer, it identified a rare CXCL13+ exhausted CD8+ T cell subset (0.8% of total T cells) as a predictor of anti-PD-L1 response, a finding missed by traditional clustering and validated via spatial transcriptomics. 25 Additionally, GCNs (Graph Convolutional Neural Networks) have been used to construct TEX regulatory networks from scRNA-seq and ChIP-seq data, prioritizing hub genes like NR4A1 that directly regulate PD-1 and LAG3—functional validation via CRISPR knockout confirmed NR4A1’s causal role in maintaining exhaustion. 26 These examples demonstrate the diverse and impactful applications of AI in ICE research, highlighting its potential to drive new discoveries and improve the accuracy and efficiency of data analysis.
While AI/ML is not an NGS technology itself, it is integral to analyzing high-dimensional NGS/multi-omics data for ICE biomarker discovery. For example, machine learning models process scRNA-seq data to classify exhausted T cell subsets with higher accuracy than traditional clustering methods. Deep learning approaches like transformers and graph neural networks (GNNs) have further enhanced utility: transformers identify context-specific exhaustion signatures by learning from thousands of single-cell profiles, while GNNs model cell-cell interactions in TME using spatial transcriptomics data. Limitations of AI/ML in NGS data analysis include dependency on large, high-quality datasets and potential algorithmic biases if training data lacks diversity. 27
3.3. Network Analysis
Network analysis is a bioinformatics approach that involves constructing and analyzing networks to understand the interactions and pathways involved in biological processes. This method is particularly useful for identifying signal transduction pathways and regulatory networks that underlie ICE. By mapping the interactions between genes, proteins, and other molecules, researchers can identify key nodes and pathways that contribute to the exhausted phenotype. 28
In the study of ICE, network analysis has been applied to scRNA-seq data to identify novel biomarkers and therapeutic targets. For instance, network construction and pathway analysis have revealed critical regulatory circuits involving inhibitory receptors like PD-1 and CTLA-4, as well as transcription factors such as TOX, which play central roles in maintaining the exhausted state of T cells.
29
These insights are crucial for designing interventions that can modulate these pathways and restore T cell function, offering new avenues for immunotherapy (Figure 2). Network analysis is frequently applied to NGS-derived data to unravel ICE regulatory pathways. Specific applications include constructing gene co-expression networks from transcriptomic data to identify hub genes (e.g., TOX, PD-1) that drive exhaustion, and mapping protein-protein interaction networks from proteomics data to reveal inhibitory receptor signaling cascades. Limitations include sensitivity to data quality and difficulty in validating predicted interactions experimentally, especially for rare cell subsets.
3.4. Proteomics and Phosphoproteomics Integration
The integration of proteomics and phosphoproteomics provides additional insights into the identification of novel biomarkers for immune cell exhaustion and the tumor microenvironment. Proteomics enables the quantitative analysis of protein expression levels within cells, while phosphoproteomics focuses on phosphorylation modifications, which are crucial for understanding cellular signaling. By integrating proteomics and phosphoproteomics data, researchers can identify protein expression and modification patterns associated with ICE, leading to the discovery of new biomarkers and therapeutic targets. 30
For example, recent studies using proteomics have revealed specific protein expression changes in exhausted T cells that are closely related to cellular dysfunction and immune suppression. Phosphoproteomics further elucidates these proteins’ phosphorylation states, providing critical clues for understanding signaling mechanisms in ICE. 31 These integrated analyses not only enhance our understanding of the molecular basis of ICE but also offer potential targets for developing new diagnostic tools and therapeutic approaches.
This multi-omics integration complements NGS data by validating transcriptional changes at the protein level and revealing post-translational modifications critical for ICE. Specific applications include quantifying PD-1/CTLA-4 protein expression in exhausted T cells and identifying phosphorylation of PI3K-AKT-mTOR pathway proteins that mediate metabolic dysfunction in exhausted T cells. Key limitations include technical variability between proteomics experiments, difficulty detecting low-abundance proteins, and the need for specialized sample preparation to preserve phosphorylation marks—making it challenging to scale for clinical samples.
3.5. Spatial Transcriptomics
Spatial transcriptomics is an emerging technology that enables the analysis of gene expression at high spatial resolution within tissue sections. Unlike traditional transcriptomics methods, spatial transcriptomics retains spatial information of cells, which is crucial for understanding cell interactions and functions within tissues. Spatial transcriptomics techniques, such as in situ hybridization and sequencing on tissue sections, can reveal the distribution and gene expression patterns of cells within tissues. 32
In the tumor microenvironment, spatial transcriptomics has been used to study the spatial distribution and functional states of immune cells. For example, studies have shown that exhausted T cells exhibit specific spatial distribution patterns within tumor tissues, which are closely related to tumor invasiveness and response to immunotherapy. 33 Additionally, spatial transcriptomics can reveal cell-to-cell interactions within the tumor microenvironment, adding a new dimension to understanding the complexity of ICE. By integrating spatial transcriptomics data with other omics data, researchers can gain a more comprehensive understanding of the molecular mechanisms of ICE and develop more effective therapeutic strategies.
As an emerging NGS-based technology, spatial transcriptomics addresses a critical limitation of scRNA-seq: lack of spatial context. Specific applications in ICE research include mapping the distribution of exhausted T cells relative to immunosuppressive TME components in NSCLC and melanoma, and identifying “exhaustion hotspots”—regions where exhausted T cells cluster with inhibitory cells—that predict immunotherapy resistance. Limitations include lower sequencing depth per cell compared to scRNA-seq, high cost of tissue processing, and limited availability of fresh/frozen clinical samples—restricting its use in large-scale studies.
3.6. Epigenetic Analysis
Epigenetic analysis is a vital tool for studying gene expression regulation, focusing on DNA modifications, histone modifications, and chromatin structure changes. Epigenetic modifications play a key role in ICE by influencing gene transcription and maintaining the exhausted state of T cells. For example, DNA methylation and histone modifications can alter gene expression levels, thereby affecting T cell function. 34
In recent years, epigenetic analysis techniques such as chromatin immunoprecipitation sequencing (ChIP-seq) and assay for transposase-accessible chromatin using sequencing (ATAC-seq) have been applied to study epigenetic changes in ICE. These techniques can identify epigenetic modification patterns associated with ICE, offering potential targets for therapeutic interventions. 35 For example, studies have found that specific histone modification patterns are closely related to the exhausted phenotype in T cells, and these modifications can regulate the expression of key genes to maintain the exhausted state. 36 Through epigenetic analysis, researchers can better understand the molecular mechanisms of ICE and develop therapies targeting epigenetic modifications. Epigenetic NGS technologies provide specific insights into the epigenetic basis of ICE. ChIP-seq applications include profiling histone modifications in exhausted T cells—for example, H3K27me3 enrichment at effector gene promoters suppresses their expression, while H3K4me3 at inhibitory receptor genes enhances transcription. 35 ATAC-seq further identifies open chromatin regions in exhausted T cells, linking epigenetic accessibility to exhaustion-related gene expression. Limitations include the need for large cell inputs that are infeasible for rare exhausted subsets, noise in low-input ATAC-seq samples, and difficulty in distinguishing cell-type-specific epigenetic changes in bulk samples.



4. Immune Cell Exhaustion and the Tumor Microenvironment
4.1. Impact of the Tumor Microenvironment on Immune Cell Exhaustion
The tumor microenvironment (TME) is a complex and dynamic milieu that exerts a profound influence on the functional state of tumor-infiltrating immune cells, with immune cell exhaustion (ICE) emerging as a key consequence of TME-mediated immune suppression. One significant factor is the upregulation of immune checkpoint ligands, such as PD-L1, on tumor cells and other cells within the TME. This upregulation interacts with inhibitory receptors on T cells, such as PD-1 and CTLA-4, leading to the suppression of T cell function and the promotion of an exhausted phenotype. 37 Additionally, the TME is rich in immunosuppressive cytokines like TGF-β and IL-10, which further inhibit T cell activation and proliferation, contributing to ICE. 38
The recruitment of regulatory immune cells, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), also exacerbates immune suppression within the TME. These cells secrete immunosuppressive factors and directly inhibit the activity of effector T cells. For example, Tregs are known to secrete TGF-β, which can suppress the cytotoxic activity of CD8+ T cells, thereby enhancing the exhaustion phenotype.39,40 Similarly, MDSCs produce arginase and nitric oxide, which inhibit T cell receptor signaling and function, promoting a state of exhaustion in T cells. 41
However, the heterogeneity of ICE within the TME complicates the identification of reliable biomarkers. The diverse phenotypes and functions of exhausted immune cells, influenced by the complex interplay of various factors in the TME, make it challenging to pinpoint specific markers that can accurately reflect the state of ICE across different tumor types and patient populations. This heterogeneity also raises concerns about the potential biases in bioinformatics analyses. For instance, the use of bulk RNA sequencing may mask the distinct characteristics of different immune cell subsets, leading to inaccurate conclusions about the overall immune status. Additionally, the reliance on predefined gene signatures may not capture the full spectrum of ICE-related changes, especially in the context of the highly variable TME.
4.2. Exhaustion Phenomena in Various Immune Cells
ICE is not limited to T cells but also affects other immune cells within the TME, including natural killer (NK) cells, B cells, and macrophages. NK cells in the TME often exhibit an exhausted phenotype characterized by reduced cytotoxicity and impaired production of cytokines like IFN-γ. This exhaustion is driven by the chronic exposure to inhibitory signals and the immunosuppressive milieu of the TME. 40 Similarly, B cells can become dysfunctional, with an altered capacity to produce antibodies and present antigens, which impacts the overall immune response against tumors. 42
Macrophages within the TME can differentiate into tumor-associated macrophages (TAMs) that support tumor growth and suppress anti-tumor immunity. These TAMs exhibit an M2-like phenotype, characterized by the production of anti-inflammatory cytokines and the promotion of tissue remodeling and angiogenesis. The interactions between TAMs and other immune cells create a complex network that fosters ICE and tumor progression. 41 Understanding these complex interactions is crucial for developing therapeutic strategies that can effectively target and reverse ICE within the TME.
Given the complexity of ICE and its impact on multiple immune cell types, it is essential to consider the potential biases in bioinformatics analyses when studying these phenomena. Single-cell RNA sequencing may offer a more detailed view of the heterogeneity within immune cell populations, but it also comes with its own challenges, such as high costs and the need for sophisticated computational tools. Moreover, the integration of multi-omics data, including proteomics and metabolomics, could provide a more comprehensive understanding of ICE, but it requires careful consideration of data integration biases and the biological relevance of the findings.
4.3. Immune Cell Exhaustion in Cancers
In colorectal cancer (CRC), T cell exhaustion is driven by TME factors including TGF-β secretion from Tregs and MDSCs, and PD-L1 overexpression on cancer-associated fibroblasts (CAFs). 34 B cell exhaustion in CRC is characterized by reduced IgA secretion and upregulation of TIM-3, impairing anti-tumor antibody responses.
NSCLC exhibits high levels of exhausted CD8+ T cells with co-expression of PD-1, LAG3, and TIGIT, particularly in smokers and patients with KRAS mutations. 43 Spatial transcriptomics has revealed that exhausted T cells cluster with M2-like TAMs in peritumoral regions, where lactate accumulation from glycolytic tumor cells further reinforces exhaustion. 44
In triple-negative breast cancer (TNBC), T cell exhaustion is driven by IL-10 and VEGF in the TME, with high TOX expression in TILs predicting resistance to paclitaxel-based chemotherapy. HNSCC is characterized by exhausted CD4+ T cells expressing CTLA-4 and PD-1, with long non-coding RNA (lncRNA) HOTAIR mediating epigenetic silencing of effector genes. In HER2-positive breast cancer, trastuzumab treatment can induce PD-L1 upregulation on tumor cells, exacerbating T cell exhaustion and highlighting the need for combination ICI therapy.
5. Identification of Novel Biomarkers
5.1. Transcriptomics and Gene Expression Analysis
Transcriptomics and gene expression analysis are crucial for identifying novel biomarkers associated with ICE. By examining transcriptomic data, researchers can identify genes that are differentially expressed in exhausted T cells compared to their non-exhausted counterparts. For instance, single-cell RNA sequencing (scRNA-seq) has been instrumental in revealing the complexity of T cell exhaustion states and identifying key exhaustion-related genes such as PDCD1, TOX, and LAG-3.45,46 These genes are consistently upregulated in exhausted T cells, providing potential targets for therapeutic intervention. 47 Gene expression profiling has also enabled the discovery of unique transcriptional signatures that distinguish exhausted T cells, offering insights into the mechanisms driving T cell dysfunction and potential biomarkers for monitoring immune responses.7,20
The application of gene expression profiling not only facilitates the discovery of biomarkers but also reveals biological findings, as exemplified by studies on various cancer types. In colorectal cancer, genes such as TOX and HIF1A, whose expression correlates with poor clinical outcomes, are potential biomarkers that can be tested via colorectal tissue biopsy or circulating tumor RNA detection, have transnational potential due to their consistent correlation with clinical outcomes across populations, and can be validated in large colorectal cancer cohorts. 34 In intrahepatic cholangiocarcinoma, the long non-coding RNA HOTAIR, which is highly expressed in exhausted CD8+ T cells, is a potential regulatory biomarker whose high expression is significantly associated with patient non-responsiveness to anti-PD-1 therapy, and it can be tested via tumor tissue biopsy or circulating RNA detection, has transnational potential for predicting therapy response, and can be validated in large cohorts. 48 In melanoma, the TOX/LAG3 gene signature is a biomarker that stratifies patients into responders and non-responders to ipilimumab/nivolumab combination therapy, and it can be tested via tumor biopsy or liquid biopsy, has transnational potential for patient stratification, and can be validated in large melanoma cohorts. 49
5.2. Epigenetic Markers
Epigenetic modifications play a pivotal role in the regulation of gene expression and the maintenance of ICE. These modifications include DNA methylation, histone modifications, and chromatin remodeling, which can stably alter gene expression without changing the underlying DNA sequence. Identifying epigenetic markers associated with ICE can provide valuable insights into the mechanisms driving this dysfunctional state and reveal new therapeutic targets. For instance, the transcription factor TOX has been identified as a key regulator of T cell exhaustion through its role in chromatin remodeling and the establishment of an exhaustion-specific epigenetic landscape. 46
Methods for identifying epigenetic markers include chromatin immunoprecipitation followed by sequencing (ChIP-seq) and assay for transposase-accessible chromatin using sequencing (ATAC-seq). These techniques allow for the mapping of histone modifications and open chromatin regions, respectively, providing a comprehensive view of the epigenetic changes associated with T cell exhaustion. Recent studies have used these methods to uncover novel epigenetic markers that are enriched in exhausted T cells, such as specific histone modifications and DNA methylation patterns.50,51 These findings highlight the potential of targeting epigenetic modifications to reinvigorate exhausted T cells and improve responses to immunotherapy. 52
DNA Methylation: DNA methylation, particularly in the promoter regions of genes, can lead to transcriptional silencing. Studies have shown that hypermethylation of the promoters of genes such as PRDM1 and BCL6 is associated with T cell exhaustion in chronic viral infections and cancer. 53 Targeting these methylation patterns with demethylating agents could potentially reverse the exhausted state.
Histone Modifications: Histone modifications, such as acetylation and methylation, can affect chromatin structure and gene accessibility. Research has identified specific histone marks, such as H3K27me3 and H3K4me3, that are differentially enriched in exhausted T cells compared to functional T cells. 36 These marks can be targeted by histone-modifying enzymes to modulate gene expression.
Chromatin Remodeling: Chromatin remodeling complexes, such as the SWI/SNF complex, play a crucial role in maintaining the epigenetic landscape of T cells. Disruptions in these complexes have been linked to the development of T cell exhaustion. 54 Targeting chromatin remodeling factors could provide a novel strategy for reversing exhaustion.
5.3. Proteomics and Metabolomics Markers
Proteomics and metabolomics offer additional layers of information that can be crucial for identifying novel biomarkers of ICE. Proteomic analysis involves the large-scale study of proteins, including their expression levels, modifications, and interactions. This approach can identify protein markers that are differentially expressed in exhausted T cells, providing insights into the functional state of these cells and potential therapeutic targets.7,45 For instance, studies have identified proteins such as Galectin-9 and TIM-3 that are upregulated in exhausted T cells and serve as biomarkers for T cell dysfunction. 55
Metabolomics, the study of small molecule metabolites within cells and tissues, can also reveal biomarkers associated with ICE. Metabolic reprogramming is a hallmark of T cell exhaustion, and specific metabolic pathways are often altered in exhausted cells. For example, the accumulation of certain metabolites and changes in metabolic enzyme expression can serve as indicators of T cell exhaustion. 56 These metabolic changes not only reflect the functional state of the cells but also provide potential targets for therapeutic intervention.19,57
Proteomics: Advanced proteomic techniques, such as mass spectrometry (MS) and liquid chromatography-tandem mass spectrometry (LC-MS/MS), enable the identification and quantification of thousands of proteins in a single experiment. These methods have been used to identify key proteins involved in T cell exhaustion, including those involved in the PI3K-AKT-mTOR pathway, which is critical for T cell metabolism and function. 56 For example, studies have shown that the expression of proteins such as PD-1, CTLA-4, and LAG-3 is significantly upregulated in exhausted T cells, providing potential targets for immunotherapy. 58
Metabolomics: Metabolomic studies have identified specific metabolic signatures associated with T cell exhaustion. For instance, the accumulation of lactate and the depletion of amino acids such as glutamine and arginine are common in exhausted T cells. 59 These metabolic changes can be targeted by metabolic inhibitors to restore T cell function. Additionally, the expression of metabolic enzymes such as IDO1 and ARG1, which are involved in tryptophan and arginine metabolism, respectively, has been linked to T cell exhaustion and immunosuppression. 59
6. Challenges in Biomarker Discovery
6.1. Data Integration and Analysis of High-Throughput Data
In the field of biomarker discovery, integrating multi-omics data and processing high-throughput data represent two critical challenges. The multi-omics approach, which combines genomics, transcriptomics, proteomics, and metabolomics data, necessitates sophisticated computational tools and algorithms for effective integration and analysis. However, the high dimensionality and variability of these datasets can lead to issues with data normalization, alignment, and interpretation, making it difficult to identify consistent biomarkers across different studies.2,5 Additionally, the processing and analysis of high-throughput sequencing data, such as that obtained from next-generation sequencing (NGS), face challenges due to the sheer volume and complexity of the data. 60 This requires substantial computational resources and sophisticated algorithms to manage data storage, processing, and analysis efficiently. 61
Furthermore, the limitations of current data analysis methods and tools pose another hurdle. Many bioinformatics tools are not fully equipped to handle the complexity of multi-omics data, often resulting in incomplete or biased interpretations. 62 The application of big data in biomarker research is promising but also fraught with difficulties. While big data approaches enable comprehensive analysis of large-scale datasets to uncover novel biomarkers and therapeutic targets, the integration of big data from multiple sources introduces challenges related to data heterogeneity, standardization, and interoperability. 63 Moreover, the interpretation of results from big data analyses requires careful consideration of the biological context and potential confounding factors, which can complicate the identification of true biomarkers. 64 Therefore, the development of advanced computational tools and algorithms capable of handling large-scale data and extracting meaningful patterns is essential. 65
6.2. Clinical Application Challenges
Translating biomarkers from research to clinical applications involves several challenges, including issues with biomarker validation and standardization. Biomarkers identified in research settings often require extensive validation in clinical trials to ensure their reliability and reproducibility. This process is time-consuming and costly, often requiring large patient cohorts and multiple validation steps. Additionally, the lack of standardized protocols for biomarker measurement and analysis can lead to variability in results, further complicating the clinical implementation of novel biomarkers. 18
Another significant challenge is the translation of biomarkers into clinically actionable tools. Biomarkers must demonstrate clear clinical utility, such as improving patient outcomes or guiding treatment decisions, to be adopted in clinical practice. 60 This requires not only robust clinical evidence but also the development of reliable and cost-effective diagnostic assays. Moreover, regulatory approval processes for new biomarker-based diagnostics can be stringent and lengthy, posing additional barriers to clinical application. 61
6.3. Complexity and Dynamic Heterogeneity of Biological Systems
The complexity and dynamic heterogeneity of biological systems add another layer of challenge to biomarker discovery. Biological systems are characterized by intricate networks of interactions among genes, proteins, and metabolites, which can vary significantly across different individuals and over time. This dynamic nature means that biomarkers identified at one time point may not be relevant or effective at another, necessitating longitudinal studies to capture the temporal changes in biomarker expression. 66
Moreover, the heterogeneity within biological systems means that a biomarker that is effective in one subset of patients may not be universally applicable. For example, immune cell exhaustion can manifest differently in various types of cancer or in different stages of the disease, requiring the identification of biomarkers that are specific to particular contexts. 67 This calls for the development of personalized medicine approaches that take into account individual differences and the dynamic nature of disease progression.
Addressing these challenges requires a multifaceted approach, including the development of more sophisticated computational models that can account for the dynamic interactions within biological systems, the use of longitudinal and multi-cohort studies to capture temporal and individual variability, and the integration of diverse data types to provide a more comprehensive understanding of disease mechanisms.
7. Case Studies and Applications
7.1. Applications in Cancer Immunotherapy
In the realm of cancer immunotherapy, identifying biomarkers associated with immune checkpoint inhibitor (ICI) therapy has been crucial for predicting patient responses and improving treatment outcomes. Research has shown that the expression of inhibitory receptors such as PD-1 and CTLA-4 on exhausted T cells correlates with responses to ICIs, and gene expression profiling has identified specific subsets of T cells, such as those expressing the transcription factor TOX, which are associated with better therapeutic responses. 43 These biomarkers help stratify patients and tailor personalized treatment plans, enhancing the precision and efficacy of cancer immunotherapy.
Additionally, applications of bioinformatics in other cancer immunotherapies, such as CAR-T cell therapy, have been significant. By using bioinformatics tools to analyze transcriptomic and epigenetic data, researchers have identified key regulators of T cell exhaustion that can be targeted to improve the persistence and functionality of CAR-T cells. 68 The integration of bioinformatics approaches has facilitated the discovery of novel biomarkers and therapeutic targets, providing new avenues for enhancing the effectiveness of immunotherapies and overcoming resistance mechanisms. 69 Moreover, in breast cancer, bioinformatics analysis of TCGA data identified Interleukin 27 Receptor Alpha (IL27RA) as a key exhaustion-related biomarker: high IL27RA expression in TILs correlates with resistance to atezolizumab, while IL27RA knockdown restores T cell effector function in preclinical models. 70 In head and neck squamous cell carcinoma (HNSCC), machine learning models integrating scRNA-seq and clinical data have developed a 6-gene exhaustion signature that predicts response to pembrolizumab with 83% accuracy. 71 These examples demonstrate how ICE-related biomarkers enable precise patient stratification and personalized immunotherapy selection.
7.2. Applications of Spatial Transcriptomics in Tumor Microenvironment Studies
Techniques of spatial transcriptomics have revolutionized the study of the tumor microenvironment (TME) by providing spatially resolved gene expression data that reveal the intricate interactions between immune cells and tumor cells. This approach has enabled the detailed mapping of immune cell distributions within tumors, shedding light on the spatial dynamics of immune evasion and the roles of different immune cell subsets in the TME. 72 By combining spatial transcriptomics with other omics data, researchers can gain a comprehensive understanding of the TME and identify novel targets for therapeutic intervention.
Studies using spatial transcriptomics have shown that the proximity of exhausted T cells to immunosuppressive cells, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), significantly impacts their functionality and response to treatment. 73 These findings underscore the importance of spatial context in immune cell interactions and highlight the potential of spatial transcriptomics to inform the development of more effective immunotherapies. .
8. Strategies to Reverse Immune Cell Exhaustion
8.1. Novel Therapeutic Strategies
ICE represents a major barrier to effective anti-tumor and anti-viral immunity, and multiple therapeutic strategies have been developed to reverse this dysfunctional state. These approaches can be broadly categorized into immune checkpoint modulation, adoptive cellular therapy, nanotechnology-mediated immune reprogramming, and T cell metabolic regulation, each targeting distinct mechanisms of exhaustion.
The development of new immune checkpoint inhibitors has been pivotal in reversing T-cell exhaustion, a state often induced by chronic infections and cancer. 74 Immune checkpoint blockades, such as those targeting CTLA-4 and PD-1, have shown significant success in reinvigorating tumor-infiltrating T lymphocytes, leading to improved clinical outcomes in cancer patients. 75 Additionally, combining checkpoint inhibitors with other treatments, such as small molecules or vaccine therapy, has further enhanced their efficacy by targeting multiple mechanisms of T-cell dysfunction.76,77
CAR-T cell therapy, another promising approach, involves reprogramming T cells to better recognize and attack cancer cells, showing substantial potential in treating hematological malignancies. 78 This therapy has been particularly effective in cases where traditional treatments have failed, offering new hope for patients with refractory cancers. 79
Moreover, the innovative use of nanotechnology has demonstrated effectiveness. Selenium nanoparticles reverse NK cell exhaustion by targeting mitochondria, reducing HLA-E expression, and enhancing immunotherapy against metastatic tumors. 80
Furthermore, targeting the metabolic pathways of T cells presents another avenue to reverse exhaustion. Altering the metabolic state of T cells can restore their function, providing a novel strategy for the treatment of chronic viral infections and cancer. 50 This approach focuses on reprogramming the energy production processes within T cells to sustain their activity and improve their ability to combat diseases. 77
8.2. Role of Bioinformatics in Therapeutic Strategies
Bioinformatics tools have become indispensable in designing and optimizing treatment plans to reverse ICE. These tools facilitate the analysis of large datasets to identify potential therapeutic targets and optimize drug combinations. 81 By leveraging high-throughput genomic tools, such as CRISPR screening and single-cell RNA sequencing, researchers can pinpoint key mediators of T-cell exhaustion and develop more targeted therapies. 82
Personalized medicine applications are particularly enhanced by bioinformatics, as they allow for the tailoring of treatments based on individual genetic profiles, thereby improving the efficacy and minimizing the side effects of immunotherapies. 83 For instance, identifying specific genetic markers associated with ICE can help in selecting the most effective checkpoint inhibitors or other immunotherapies for individual patients. 64 The integration of bioinformatics with clinical data enables the continuous refinement of therapeutic strategies, ensuring that treatments remain effective as new insights into ICE emerge. 55
Moreover, bioinformatics tools play a critical role in the discovery and validation of novel biomarkers that predict patient responses to therapies. This is essential for developing more precise and effective treatment plans that can adapt to the dynamic nature of immune responses in patients with chronic infections or cancer. 81 By combining bioinformatics with experimental data, researchers can accelerate the development of new therapies and improve the management of immune-related diseases. 82
8.3. Real-Time Dynamic Monitoring of Treatment Responses
A promising area where bioinformatics can significantly contribute is in the real-time dynamic monitoring of treatment responses and drug resistance evolution. Advances in technology now allow for the continuous collection of live body fluid data, such as circulating tumor DNA (ctDNA) and immune cell profiles from blood samples. Bioinformatics tools can analyze these data in real-time to track the efficacy of treatments and the emergence of drug resistance. 84
For example, by monitoring changes in ctDNA levels, researchers can assess the response of tumors to immunotherapy and detect early signs of resistance. Similarly, analyzing immune cell profiles can provide insights into the immune system’s dynamic response to treatment, allowing for timely adjustments in therapeutic strategies. 26 This real-time monitoring can help in the early identification of patients who are not responding to treatment, enabling clinicians to switch to alternative therapies promptly.
8.4. Predictive Modeling of Treatment Outcomes
Bioinformatics can also be used to develop predictive models that forecast patient outcomes based on initial treatment responses and genetic profiles. Machine learning algorithms can integrate data from multiple sources, including genomic, transcriptomic, and clinical data, to generate personalized treatment plans. These models can predict the likelihood of treatment success and the potential for drug resistance, guiding clinicians in making informed decisions. 85
For instance, AI-driven models can analyze the expression patterns of exhaustion markers and other relevant genes to predict patient responses to immune checkpoint inhibitors. By incorporating real-time data from live body fluid monitoring, these models can be continuously updated to reflect the current state of the patient’s disease and immune response. 86
8.5. Future Directions
The integration of bioinformatics with real-time monitoring and predictive modeling holds great promise for the future of personalized medicine. As technologies continue to advance, the ability to collect and analyze live body fluid data in real-time will become more accessible and cost-effective. This will enable the development of more sophisticated models that can adapt to the dynamic nature of immune responses and disease progression. 87
Moreover, the combination of bioinformatics with emerging technologies such as liquid biopsies and wearable devices will provide a comprehensive view of patient health. These integrated approaches will not only improve treatment outcomes but also enhance patient quality of life by reducing the need for invasive procedures and enabling early intervention. 88
9. Conclusion and Future Directions
In conclusion, bioinformatics has significantly advanced the understanding of ICE by identifying key molecular pathways and gene expression profiles. 82 Key findings indicate that T cell exhaustion is marked by the upregulation of inhibitory receptors and significant metabolic alterations. 50 Despite these advancements, challenges remain, such as the heterogeneity of exhausted immune cells and the difficulty in targeting these cells without affecting overall immune function.89,90
Future research should focus on developing advanced bioinformatics tools and techniques, For instance, the integration of multi-omics data, including genomics, transcriptomics, proteomics, and metabolomics, could provide a more comprehensive understanding of the molecular mechanisms underlying T cell exhaustion. These tools will be crucial in biomarker identification, leading to more effective and personalized treatment strategies. 18 Additionally, exploring spatial transcriptomics could offer insights into the spatial organization and interactions of T cells within their microenvironment. This technology could reveal how the local environment influences T cell exhaustion and identify potential targets for localized therapies. 91
The identification and validation of biomarkers hold significant clinical implications, enhancing diagnosis and treatment by providing insights into patients’ immune status and predicting their response to therapies. 92 Integrating biomarker data with patient-specific genetic information can help tailor immunotherapies, improving outcomes and reducing adverse effects. 93 Developing robust bioinformatics tools to analyze and interpret biomarker data will be essential in advancing immunotherapy. Additionally, a phase II trial in HNSCC (NCT04524871) is currently testing a Transmembrane Protein 71 (TMEM71)-targeted vaccine to reverse T cell exhaustion, with preliminary data showing restored IFN-γ production in 67% of patients. To accelerate clinical translation, future efforts should focus on validating candidate biomarkers through multi-center, prospective trials across diverse cancer types while developing standardized assays for reliable biomarker detection. These biomarkers should be integrated into clinical decision-support tools, including AI-driven algorithms that can predict both ICI response and IRAE risk, and researchers should explore combination strategies targeting pathways identified through bioinformatics to reverse ICE. Concurrently, regulatory frameworks for companion diagnostics must be updated to accommodate multi-omics-derived biomarkers, ensuring their timely approval and widespread accessibility to patients and healthcare providers.

Footnotes
Author Note
The authors utilized ChatGPT-4.0 to assist with language refinement during the preparation of this work. All content was subsequently reviewed and revised by the authors, who take full responsibility for the final version of the publication.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study design, data collection, data analysis, manuscript preparation, and publication decisions of this work were supported by Zhejiang Provincial Natural Science Foundation (No. LQN25H270011 by YH), Zhejiang Province Medical and Health Science and Technology Project (No. 2024KY1201 by YH).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.