
Abstract
Lymphoma is a highly heterogeneous malignancy, demanding accurate and precise diagnosis to guide the selection of the appropriate treatment for optimal outcome. Copy number aberration (CNA) has been suggested to play an important role in the occurrence and development of lymphoma and thus can be explored as biomarker to improve disease management. It is believed that CNAs in variable forms and complexities can be triggered by both exogenous (eg viral infection and ionizing radiation) and endogenous factors (eg genetic predisposition and evolutionary forces). However, conventional cytogenetic methods have limitations to detect all types of CNAs with accuracy and adequate details. The emergence of new technologies, including fluorescence in situ hybridization (FISH), chromosome microarray analysis (CMA), and especially next-generation sequencing (NGS) has made significant progress in the identification and characterization of CNAs or CNA-related genomic aberrations. Accumulating data addressing molecular insights and clinical implications have provided us more theoretical and experimental support for its clinical translation. Currently, while only limited number of CNAs or CNA-related genomic variation, such as deletion/amplification of DNA segments, have been documented in major guidelines or consensus for their clinical potential in lymphoma, more CNAs remain to be further characterized and/or discovered for their clinical relevance. Taking together, with available and upcoming evidence, CNA should play an important role as a diagnostic and prognostic biomarker while integrated with the current settings in lymphoma.
Keywords
Introduction
Lymphoma is a malignancy that originates in lymphoid hematopoietic tissue. In 2022, the incidence number of lymphoma was approximately 635,000 in the world, according to the International Agency for Research on Cancer. 1 Lymphoma can be divided into Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL) by pathological features. Based on the fifth edition of the WHO Classification of Haematolymphoid Tumours (WHO-HAEM5), lymphomas are classified into 125 entities/types by a grading system. 2 Accurate and precise classification and diagnosis of lymphoma can help guide the selection of the appropriate treatment for optimal survival. At present, the diagnosis and classification of lymphoma generally follow clinical manifestation, morphologic criteria, immunophenotype, and genetic features. 3
Genetic features have been the subject of extensive research in the field of cancer diagnostics in recent years. A wide range of molecular techniques have been employed to detect gene mutation and chromosomal variation. 4 Considering that the genetic mutations present among lymphoma entities display great heterogeneity, methods such as polymerase chain reaction (PCR), Sanger sequencing, and next-generation sequencing (NGS) are widely used for screening mutated genes in order to classify and risk-stratify lymphomas. 5
In addition to gene mutation, the importance of chromosomal variation is increasingly being recognized. Chromosome variation mainly includes chromosome numerical abnormality and structural variation (SV), which often leads to copy number aberration (CNA) of DNA segments or involved genes in the genome.6,7 Although CNAs may occur less frequently than other types of genetic variations, such as single nucleotide variants (SNVs) and small insertions and deletions (InDels), they are more likely to influence the phenotypes due to their long regions of variation. 8
With the increase of research on CNA, the application of CNA in lymphoma has been included in relevant guidelines and standards. Guidelines or consensus such as WHO-HAEM5, National Comprehensive Cancer Network (NCCN), and American College of Medical Genetics and Genomics and the Cancer Genomics Consortium (ACMG/CGC) guidelines, have all indicated that CNAs or CNA-related genomic aberrations are involved in the classification, disease transformation, and prognostic assessment of lymphoma entities.2,9 Studies have demonstrated that the presence of CNAs may play a key role in the occurrence and progression of lymphoma, such as diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), et al10–12 Therefore, it is necessary to detect CNA and explore its biological insights and clinical significance.13,14
In this paper, we provide a comprehensive review of the current understanding of CNA formation and pathomechanisms, as well as recent advances in CNA detection in lymphoma. We further emphasize the clinical significance of CNAs, including those strongly endorsed by major guidelines and consensus statements, as well as those supported by high-impact studies. This review aims to advance the application of NGS technologies for high-resolution, genome-wide CNA analysis, facilitating integration with existing cytogenetic and molecular diagnostic data to enhance precision medicine in lymphoma.
Formation and Pathomechanisms of CNA
CNAs in human chromosomes were reported as early as in the 1960s.
15
In recent years, CNA-related research has increased rapidly, resulting in a gradual understanding of CNA formation and pathogenicity.
16
To date, it is generally believed that both exogenous triggers, such as viral infection and ionizing radiation (IR), and endogenous processes, such as genetic predisposition and evolutionary forces, are the main risk factors involved in CNA formation

Risk Factors and Underlying Mechanisms of the Formation of CNA. (
It has been reported that 12% of human cancers worldwide are attributed to viruses, which integrate into the host genome, leading to genomic instability and dysregulation of cancer related genes. 23 Taking gastric cancer (GC), for example, helicobacter pylori infection of the stomach impairs DNA repair factors, leading to the accumulation of DNA damage, exhibiting the genomic characteristics of gastric adenocarcinoma associated with copy number amplification. 24 Epstein-Barr virus (EBV) infection is believed to be closely associated with the occurrence of various lymphomas, such as Natural killer/T cell lymphoma (NKTCL), Burkitt lymphoma (BL), and HL. EBV infection leads to susceptibility of NK or T cells to other genomic aberrations, which is an early event of NKTCL. 25
As one of the endogenous processes of CNA formation, germline mutations in susceptibility genes may contribute to cancer susceptibility through biological processes such as DNA repair and cell cycle regulation. 6 Similar to Darwinian evolution, the development of cancer can be based on the continuous accumulation of random mutations in cells, which confers on the cells the ability to proliferate more efficiently, eventually enabling them to proliferate without limit, invade tissues, and form cancer. 26
DNA damages caused by IR may result in DNA single-strand break (SSB) or DNA double-strand break (DSB). 17 It has been shown that IR-based radiotherapy can induce DNA damages that contribute to the poor outcome in cancer patients in a study conducted in 190 paired primary and recurrent gliomas from the Glioma Longitudinal Analysis (GLASS) Consortium and 3693 post-treatment metastatic tumors from the Hartwig Medical Foundation. 27 IR-induced DNA damage activates the DSB repair mechanism, and the failure in proper DSB repairment may lead to the genomic alternations that can be identified as prognostic and predictive biomarkers.
The interaction of various exogenous triggers and endogenous processes leads to DNA damage and eventually evolves into somatic DNA variation
CNAs can be categorized into 5 grades: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign, according to the American College of Medical Genetics and Genomics/Association for Medical Pathology (ACMG/AMP) sequence variant interpretation guidelines. Among these 5 grades, pathogenic CNAs refer to CNAs that have been reported to be associated with a consistent clinical phenotype and contain pathogenic small CNAs or dose-sensitive genes. 30 During the occurrence and progression of diseases, pathogenic CNAs directly or indirectly lead to changes in mRNA and protein levels, or the formation of new products through the recombination of gene functional domains, thereby acquiring new functions. 16 In this regard, a large number of investigational studies have found that CNA is closely related to a number of different types of diseases, including oncological diseases, as well as autoimmune diseases, cardiometabolic diseases, and neurological/psychiatric diseases, with an important biological role in the occurrence and progression of these diseases.31,32
Methods for CNA Detection
For decades, there have been a variety of methods being developed for the detection of CNAs or genetic abnormalities associated with CNAs, and significant progresses have been made in research and clinical applications associated with relevant diseases based on that

The Project Timeline of CNA with Clinical Significance. It Includes the Time Points of the Invention of Novel CNA Detection Methods (Left) and Major Research and Clinical Progress (Right).9,33–46 Abbreviations: CML, chronic myeloid leukemia; CN-LOH, copy-neutral loss of heterozygosity; ICGC/PCAWG, International Cancer Genome Consortium/Pan-Cancer Analysis of Whole Genomes.
Advantages and Limitations of Methods for CNA Detection.

Karyotyping
As the initial method to detect CNA, the emergence of chromosome karyotyping led to the establishment of traditional cytogenetic techniques. Karyotyping involves the separation of intact chromosomes from mid-dividing or metaphase cells, followed by hypotonic treatment of the samples and observation of the chromosomes with an optical microscope.55,56 Later, the introduction of chromosome banding techniques, such as G-banding, further refined karyotyping and promoted the development of cytogenetics. 57 By chromosomal karyotyping, chromosome numbers and large structural variations (>5 Mb) can be identified.55,58 Up to now, karyotyping remains as the gold standard method for cytogenetic studies. 59 However, due to the limitation of resolution, karyotyping makes it difficult to detect subtle variations, including CNA at single-gene levels, in chromosomes. 58
FISH
FISH refers to the in situ hybridization of fluorescently labeled DNA or RNA probes to cellular targets to obtain information about chromosomes. Compared to karyotyping, FISH doesn’t have to be performed on dividing cells, thus taking less time since no cell culture is required. 60 As a diagnostic method for cancer, FISH has often been used as a complementary tool for chromosome karyotyping. 58 The NCCN guidelines proposes that molecular analysis of copy number amplification or deletion, gene rearrangement, and chromosomal translocation by FISH technique is helpful in the diagnosis of lymphoma. 61 In the case of CLL/SLL, the most common CNAs identified by the FISH technique are 13q14 deletion, 11q deletion, 12q trisomy, 17p deletion, and 6q deletion. Among them, patients with 17p deletion have the lowest survival rate. 62 Nowadays, FISH is a well-established method for chromosome research and diagnosis, comprising hundreds of probes and probe sets, including chromosome-specific repetitive DNA sequences probes, locus specific probes, and whole chromosome or chromosome region painting probes.60,63 However, the application of the standard FISH is limited to chromosomal abnormalities that have been previously identified as known targets and to the availability of the probes. Moreover, as with karyotyping, almost all FISH analysis relies on manual analysis, which can cause bias and even errors due to observer's subjectivity and variation in experience.48,64
CMA
The introduction of microarray-based methods has accelerated the development of CNA detection. 65 In 1998, comparative genomic hybridization (CGH)-based arrays (aCGH) was introduced, using microchip attached with thousands of DNA probes made up of known sequences in the genome for competitive hybridization of experimental and control DNA samples. 33 The aCGH uses DNA sequences for analysis with high efficiency and high resolution compared to karyotyping and can detect chromosome aneuploidy, microdeletions, microduplications, and unbalanced rearrangements.66,67 However, aCGH can only define regions of chromosomal imbalance and cannot detect balanced rearrangements since it obtained results by comparing the experimental sample with the control sample. 49 In hematologic tumors, CMA is used for molecular karyotyping to evaluate diagnostic and prognostic CNAs. 68
NGS
NGS technology was invented and developed rapidly in the early twenty-first century to achieve massive parallel analysis (MPS) at much lower cost and faster speed compared to Sanger sequencing. NGS mainly uses read pairs (RP), split-read (SR), and read depth (RD) to indicate copy number aberrations. 55 Commonly used types of application of NGS in cancer studies include DNA-based WGS, WES, targeted-panel sequencing, and RNA-based whole-transcriptome sequencing (WTS) or RNA sequencing. 58 With these methods, the full range of genomic abnormalities can be detected, including point mutations, small insertion/deletion, structural variants, gene fusions, gene expression, and DNA methylation, to define various genetic changes in most lymphomas.58,69,70 NGS offers superior sensitivity, detecting variants at much lower frequencies (∼1%) compared to Sanger sequencing. 71 It also identifies complex CNAs that are often missed or poorly resolved by amplicon-based methods, CMA, or FISH. 72 Additionally, NGS enables highly efficient and cost-effective testing through multiplexing, allowing simultaneous analysis of multiple samples and/or genetic targets in a single experiment. 73 In recent years, liquid biopsy based on NGS technology and noninvasive detection of circulating free DNA (cfDNA) and circulating tumor DNA (ctDNA) in cancer patients has become a hot topic in both research and clinical application.74,75 Thus, ctDNA-based minimal residual disease (MRD) monitoring has emerged as a powerful tool for detecting residual cancer post-treatment, enabling early relapse prediction, remission confirmation, and personalized treatment decisions. 76 Similarly, prior studies have shown that NGS analysis of cfDNA, assessing CNAs and fragment size, combined with the assays of tumor markers in peripheral blood, enables effective early cancer detection or MRD monitoring in cancer patients.77,78
Traditional methods like karyotyping and FISH remain common for CNA detection in cancer, but their limitations—low sensitivity, reliance on predefined targets, and inability to effectively capture tumor heterogeneity, subclonal variants, or complex CNAs—restrict their utility in modern clinical management. These methods also lack the multiplexing capacity to simultaneously analyze multiple samples, genetic targets, or diverse genetic alterations. To this end, NGS has emerged as a cost-effective alternative, offering enhanced sensitivity and comprehensive CNA detection in lymphoma and other cancers.32,79,80 However, its routine clinical adoption faces challenges, including moderate costs that must balance analytical capabilities (eg, sensitivity, complexity, and multiplexing), complex data analysis, and the need for standardized laboratory and bioinformatics protocols. Integrating CNAs with mutations and fusion genes is critical for comprehensive cancer profiling to guide precise diagnostics and therapies, yet this introduces additional hurdles in data integration, computational demands, and clinical decision-making. Future advancements, including automated bioinformatics pipelines, cost-efficient workflows, clinical trial validation, and the integration of artificial intelligence (AI), will be essential to overcome these barriers and fully harness NGS for multi-gene analysis in precision oncology.81,82
CNAs, including gains and losses of genomic regions, are significant drivers of cancer initiation, progression, and therapeutic resistance across various cancer types, including lymphoma. They contribute to genomic instability and aneuploidy, impacting gene expression and tumor evolution. 83 In lymphoma, such as classical Hodgkin lymphoma (cHL) 84 and adult T-cell leukemia-lymphoma (ATL), 85 CNAs are critical for understanding clonal architecture and identifying therapeutic targets.86,87 Single-cell RNA sequencing (scRNA-seq) uncovers intra-tumor heterogeneity, identifying rare subclones, and linking genomic alterations to clinical outcomes. The scRNA-seq highlights key CNAs in immune evasion and clonal expansion, reveals subtype-specific alterations and therapeutic targets. Despite challenges like technical noise and cost, ongoing advancements in tools and multi-omics integration promise to enhance its utility in precision oncology. 83
Diagnostic and Prognostic Potential of CNAs in Lymphoma
It was once believed by clinicians and cytogeneticists that chromosomal aberrations did not cause clinically significant alterations and were not clinically important.
88
However, George M and Yanka M discovered chromosome 14 aberration in BL, which initiated the study of chromosomal aberration in lymphoma.
34
Subsequent studies have further confirmed that recurrent CNAs were identified in lymphoma and even noted in the relevant guidelines for diagnosis and prognostic stratification.79,89 We searched the ClinicalTrials.gov registry (https://clinicaltrials.gov/) using the searching terms “lymphoma” in the Condition/Disease field and “copy number” in the Other Terms field and retrieved 8 trials

CNAs with Clinical Relevance in Lymphoma. Clinically Significant CNAs in Lymphoma are Depicted in the Diagram, Organized by Their Chromosomal Locations, with Annotations Indicating Associated Disease Indications, DNA Fragments, and Major Cancer-Related Genes Within These Regions. Green Denotes Gain or Amplification, While red Signifies Deletion or Loss.
Clinical Trials Investigating CNAs in Lymphoma.

CNA or CNA-Related Detection Indicated in Guidelines and/or Consensus in LBCL.
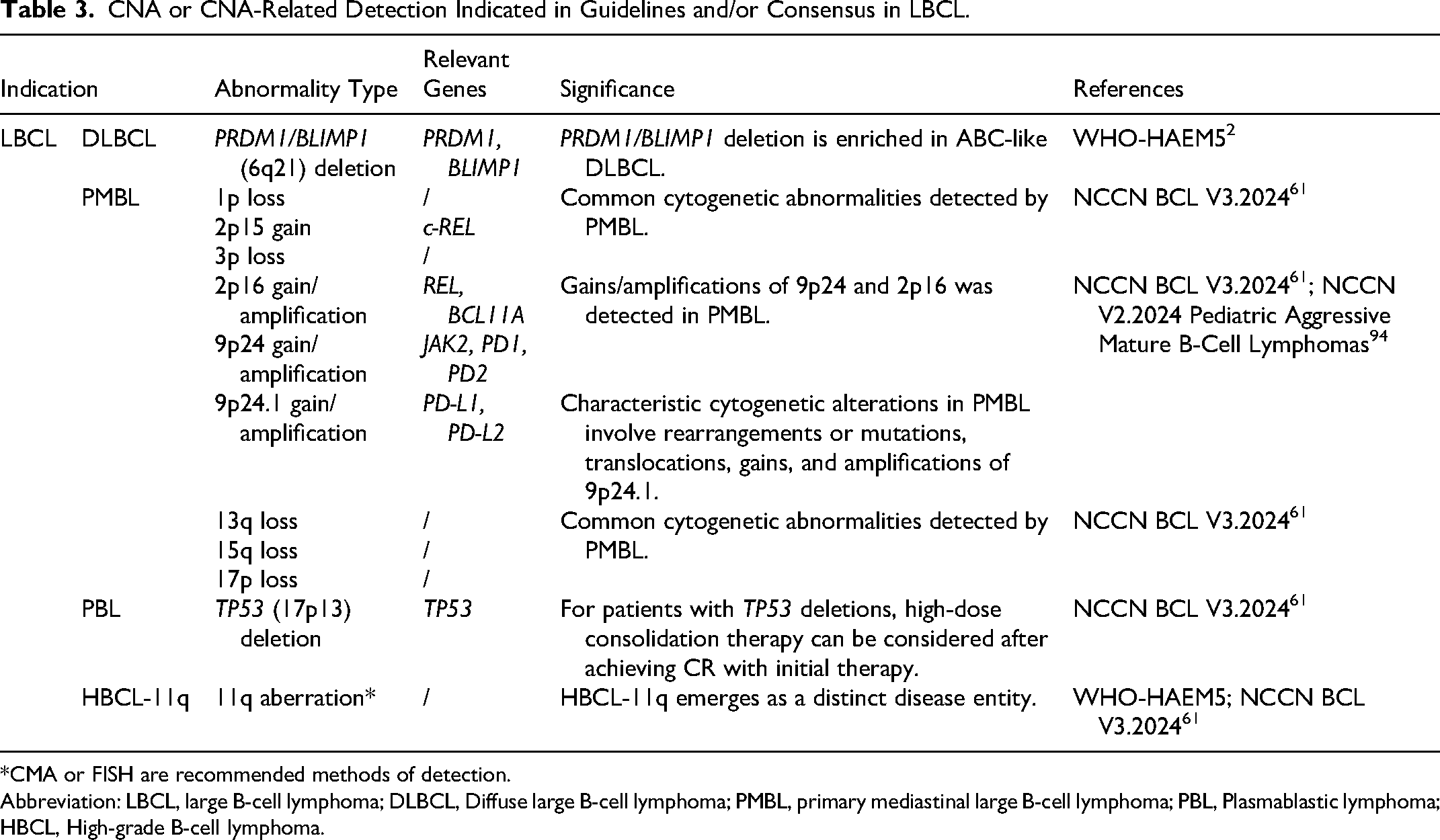
*CMA or FISH are recommended methods of detection.
Abbreviation: LBCL, large B-cell lymphoma; DLBCL, Diffuse large B-cell lymphoma; PMBL, primary mediastinal large B-cell lymphoma; PBL, Plasmablastic lymphoma; HBCL, High-grade B-cell lymphoma.
CNA or CNA-Related Detection in Guidelines and/or Consensus in CLL/SLL.

*FISH is recommended method of detection.
Abbreviation: CLL/SLL, Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma.
CNA or CNA-Related Detection in Guidelines and/or Consensus in Other B-NHL.

*Karyotype or FISH are recommended methods of detection.
Abbreviation: FL, Follicular lymphoma; MZL, Marginal zone lymphoma; NMZL, Nodal Marginal Zone Lymphoma; EMZL, extranodal marginal zone lymphoma.
CNA or CNA-Related Detection in Guidelines and/or Consensus in T-Cell and NK-Cell Lymphomas.

*Karyotype or FISH are recommended methods of detection.
Abbreviation: ALCL, Anaplastic large cell lymphomas; HSTCL, Hepatosplenic T-Cell Lymphoma; PTCL, peripheral T-cell lymphomas; T-PLL, T-Cell Prolymphocytic Leukemia; EATL, Enteropathy-associated T cell lymphoma; MEITL, Monomorphic epitheliotropic intestinal T-cell lymphoma; ENKTCL, Extranodal NK/T-cell lymphoma.
NHL
NHL is a collection of malignant neoplasms originated from B-lymphocytes, T-lymphocytes, or natural killer cells. These lymphoma entities differ in cytologic and histopathologic features, immunophenotypes and genome variation backgrounds.2,97 Significant biological heterogeneity makes lymphoma an application of precision medicine, which provides tailored diagnostic, prognostic, and therapeutic options for each patient based on integrated information about the patient's own physiological characteristics and the molecular features of the malignant cells. Among them, CNA is becoming an important factor for improving treatment selection and clinical management.5,98 Detections of CNAs or CNA-related genomic aberrations have been recommended by major guidelines and/or consensus in lymphoma
Large B-Cell Lymphoma (LBCL)
DLBCL is the most common type of LBCL
2
and a type of aggressive lymphoma with a rapid rate of disease progression.
99
First-line treatment with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) enable about 60% of patients to be cured, while remaining patients are still at risk of relapse or progressive disease.10,100 Improvements in DLBCL outcomes required a better understanding of clinical and pathologic heterogeneity. More accurate typing based on cell origin and genetic alterations allows different DLBCL patients to receive precise and individualized treatments for better outcomes.
101
Based on gene expression profiles and cell of origin (COO), DLBCLs can be classified into germinal centre B-cell-like (GCB) DLBCL and activated B-cell-like (ABC) DLBCL.
102
ABC DLBCL patients usually have worse clinical outcomes than GCB DLBCL patients after they were treated with R-CHOP, which is likely caused by a series of subtype-specific genetic abnormalities, including CNAs.103,104 In the WHO-HAEM5, it has been documented that genetic abnormalities, such as PRDM1/BLIMP1 deletion, may block the B-cell differentiation program in ABC DLBCL
It has been found that GCB DLBCLs present genetic alterations on 1p (including the gene TP73), 2p (REL), 10q (PTEN), 12q (MDM2), 13q (MIHG1, ING1). The REL gene is a member of the NF-κB transcription factor family, and amplification of the REL enhances the response of the NF-κB pathway to microenvironmental signals. 105 In GCB DLBCL, PTEN deletion tends to be accompanied by t (14, 18) and may contribute to t (14, 18)-related pathogenesis.105,106 ABC DLBCL is characterized by CNAs in trisomy 3, 3p (FOXP1), 3q (NFKBIZ), 9p (CDKN2A, CDKN2B, INK4a/ARF), 18q (BCL2, NFATC1), 19q (SPIB). 105 For example, trisomy 3 and deletion of INK4a/ARF tumor suppressor locus have been identified almost exclusively in ABC DLBCLs.105,107 Trisomy 3 and 3p (FOXP1) amplification brings about high expression of FOXP1, which has been considered as an independent poor prognosis factor for ABC DLBCL. 108 Further investigation has found that FOXP1 may interact with IRF4, SpiB-binding regions, as well as NF-κB common motifs to enhance B-cell survival. FOXP1 also activates MYC expression and IKK phosphorylation and potentiates multiple signaling pathways such as the NF-κB pathway. Based on accumulating evidence, it has been proposed to use comprehensive analysis of genetic mutations and SVs or CNAs to improve diagnostic and prognostic vale in DLBCL.100,109,110
In addition to DLBCL, CNAs have also been reported in other disease entities in LBCL. According to the NCCN guidelines, detection of common CNAs has a potential clinical value in primary mediastinal large B-cell lymphoma (PMBL) and plasmablastic lymphoma (PBL).94,111 In WHO-HAEM5, Burkitt-like B-cell lymphoma without MYC rearrangement can be identified as high-grade B-cell lymphoma with 11q aberrations (HGBL-11q), if there is an increase/deletion of chromosome 11q
CLL/SLL
CLL/SLL is the most common hematological malignancy in the western countries and features the accumulation and proliferation of mature B cells in the blood, bone marrow, and lymph nodes.12,113 The clinical course of different CLL/SLL patients varies greatly, so it is proposed to detect and analyze the genetic characteristics of patients for prognostic evaluation. 114
Regarding CNA-related genomic aberrations, the NCCN guideline suggests that del(13q), del(11q), trisomy 12, del(17p), and del(6q) are the most common abnormalities in CLL/SLL
Del(13q) are present in 55% of CLL/SLL and encompass MIR15A and MIR16-1. 79 It has been shown that sole 13q deletion had a better median treatment-free interval and PFS, whereas patients with del(11q) and del(17p) had more severe disease than others. 62 In CLL/SLL, del(11q) always involves ATM, which is associated with poor prognosis. 116 More studies have confirmed that patients with mutated TP53/del(17p) had worse PFS compared to the subgroup defined by recurrent genetic alterations.12,117
Approximately 15% of patients with CLL/SLL develop Richter's syndrome (RS), which manifests as histological transformation to DLBCL. In these patients, the most common genetic abnormalities involved TP53 deletion, trisomy 12, 13q14.3 deletion (DLEU2/MIR15A/MIR16B), and CDKN2A deletion. TP53 deletion and CDKN2A deletion define a poor survival outcome.118,119 Gains of 2p have been associated with an increased risk of Richter syndrome transformation. In CLL/SLL, MYCN has a pathogenic role, and REL gain was associated with shorter survival. The prediction of survival outcomes in patients with CLL/SLL may be significantly enhanced by assessing the presence of gains at 2p (MYCN, REL) and 8q genomic regions (MYC).120–122 In summary, significant amount of studies have identified various forms of CNAs or CNA-related genomic aberrations that may have clinical implication in CLL/SLL, which should be further investigated for their mechanistic insights and clinical translations.
Other Types of B-NHL
CNAs are involved in the classification and prognostic assessment of multiple disease entities as indicated in the WHO-HAEM5.
2
FL represents about 22% of new NHL cases and is the most common type of indolent NHL.
61
According to the WHO-HAEM5, classical FL is a subtype with a follicular growth pattern and accompanied by t(14;18) (q32;q21) causing BCL2 overexpression, which is distinguished from the other two subtypes of FL: follicular large B-cell lymphoma (FLBL) and FL with uncommon features (uFL).
2
Notably, BCL2 amplification, which may also result in BCL2 overexpression, is less common and is not recommended by major guidelines for routine diagnostic evaluation in FL. In the 2024 NCCN, a rare FL subtype with a good prognosis is also described, which is usually lack of t(14; 18), but in the presence of STAT6 mutations, 1p36 deletion
Studies have identified chromosomal abnormalities associated with FL, including frequent copy number gains, such as 1q, 2p, 6p, 7, 9q, 12q, 17q, 18, and X, and frequent copy number losses, such as 1p36, 6q, 10q23.1-q25.1 (FAS, PTEN), and 17p (TP53).123–125 It has also been shown that gain of X or Xp and losses of 6q23.2–24.1 or 6q13–15 were correlated with poor overall survival (OS). 124 The loss of TNFRSF14 (1p36.32) increases BCR signaling, leading to autonomous activation of B cell proliferation and changes in the tumor microenvironment, thereby driving disease progression in FL. 126 Deletions in CREBBP (16p), TP53 (17p), and CDKN2A/B (9p) have been identified to be associated with inferior outcomes.125,127 A study of FL has identified two regional alternations (del(1) 1p36.22-p36.33 and del(6) 6q21-q24.3) that are risk predictors of transformation, which are independent of the International Prognostic Index (IPI) and associated with a high-risk of transformation and inferior outcome. 123 In another study, it has been found that abnormalities of 3q27.3-q28, chromosome 11, 9p21.3, and 15q were more common in tFL than in FL samples. These CNAs may cause disruption of the p53 pathway, activation of NF-κB, dysregulation of transcription factors, and immune evasion. 124
In marginal zone lymphoma (MZL), the common CNAs were different among subtypes, including extranodal marginal zone lymphoma (EMZL) of mucosa-associated lymphoid tissue, nodal marginal zone lymphoma (NMZL), and primary cutaneous marginal zone lymphoma (PCMZL). While gain of chromosome 2p and 6p and loss of chromosome 1p and 6q are frequent in NMZL, 6p gain and 6q loss are only seen in EMZL of the ocular adnexa
In mantle cell lymphoma (MCL), it has been shown that CNAs, including TP53 alterations and loss of 9p21/CDKN2A, 13q14/RB1, and 9q22-q31, are associated with poorer survival. 129 This study also found that alterations of 15q14-q21, 8q24/MYC, SP140, 18q21-q22, and 13q33-q34 were also found to be significantly associated with shortened OS. Additional study has suggested that CNAs have significant prognostic implications, including the presence of TP53 (17p) deletion and mutations, CDKN2A (9p21) deletions, as well as CDK4 amplification, and recommended FISH for TP53 or deletion of 17p in the diagnosis of MCL. 130
T-Cell and NK-Cell Lymphomas
CNAs are also present in T-cell non-Hodgkin lymphomas. It is proposed in the NCCN Guidelines of T-cell lymphomas (NCCN-TCL) that isochromosome 7q and trisomy 8, which are associated with 7p loss and amplification of 7q, are the most common chromosomal abnormalities in hepatosplenic T-cell lymphoma (HSTCL).
131
The guidelines also indicate that CNAs should be examined as potential markers, including ATM deletion in T-PLL, 9q34 gain and 16q12 loss in EATL and MEITL, and 6q21–25 deletion in extranodal NKTCL
CDKN2A deletions (9p21) show subtype-specific prevalence in cutaneous TCL, occurring frequently in mycosis fungoides and Sézary syndrome (>40%) but rarely in CD30+ cutaneous anaplastic large cell lymphoma. 132 In peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), CDKN2A losses are common (46%) and are associated with inferior PFS and OS. 133 In contrast, T-ALL/LBL cases with CDKN2A deletions (55% prevalence) exhibit improved treatment response, prolonged OS, and enhanced MRD clearance post-induction. 134
Analysis of NGS data revealed that the 14q11.2 region, which contains the T-cell receptor (TCR) gene, is deleted in most T-cell lymphomas, but rarely the other types of lymphomas. This makes it possible to identify T-cell lymphomas by detecting CNAs in 14q11.2. 135 Integrated with DNA methylation, genome-wide CNA profiling has been employed for dividing adult T-cell leukemia/lymphoma (ATL) into subtypes: T-cell lymphoblastic leukemia (T-ALL) and T-cell lymphoblastic lymphoma (T-LBL). 136 Similarly, nodal EBV+ T/NK-cell lymphoma has been identified as a subtype of NKTCL that is associated with different CNAs. Nodal EBV+ T/NK-cell lymphoma is characterized with deletion of 14q11.2, which is correlated with loss of TCR loci and T-cell origin, suggesting that 14q11.2 deletion may potentially be a useful diagnostic biomarker. 137 In cutaneous T-cell lymphoma (CTCL), 7q21.3 amplification drives ectopic expression of PEG10, thereby achieving therapeutic resistance via the PEG10/KLF2/NF-κB pathway. 138
HL
HL is categorized into nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) and classic Hodgkin lymphoma (cHL). The cHL accounts for approximately 90% of HL and is characterized by the presence of Hodgkin Reed-Sternberg (HRS) cells in an inflammatory background. 139
Multiple studies have identified focal copy number gains and losses that play a role in the development and progression of cHL. Alteration of protein programmed death 1 ligand 1 (PD-L1)/ protein programmed death 1 ligand 2 (PD-L2) caused by copy number aberration of 9p24.1 is a typical feature of HL. 140 HRS cells frequently manifest copy number amplification at 9p24.1, which contains the genes CD274 and PDCD1LG2, encoding PD-L1 and PD-L2, respectively. 141 PD-L1 gene amplification increases protein expression and activity, and subsequent binding to the PD-1 receptor on T cells that leads to the inhibition of T cell activation, thereby protecting HRS cells from T-cell-mediated killing. 142 The JAK2 gene is also located in chromosome 9p24.1. The 9p24.1 amplification may also lead to increased JAK2 expression and elevated JAK/STAT signaling, which further induces PD-1 ligand expression. 143 A previous study has shown that patients with early-stage cancer has less 9p24.1 amplification and a favorable outcome compared to those with advanced diseases, thus 9p24.1 alterations can be exploited for predicting the prognosis of cHL patients. 144 In consistent with this finding, early stage cHL has a unique sensitivity to PD-1 blocking antibodies, such as nivolumab and pembrolizumab. 145 Similarly, a study has found that 9p24.1 alterations and PD-L1 expression can be explored as a biomarker to predict the outcome in patients with cHL, showing less responses to PD-1 blockade and inferior outcomes in patients with lower levels of 9p24.1 alterations and less PD-L1 expression on HRS cells than those with higher levels of 9p24.1 alterations and higher PD-L1 expression. 146
Via liquid biopsy, recurrent CNAs in cHL were identified by targeted cfDNA sequencing, including frequent copy number gains such as 19p13.2, 2q31.2, 12q13.3 (STAT2, STAT6), 2p16.1 (REL), 9p24.1 (JAK2, CD274) and frequent copy number losses including 6q23.3 (TNFAIP3, ECT2L), 9q13, 13 q32.3, 6 q22.31 and 15q15.1. 147 It has been demonstrated that other CNAs may also impact the prognosis or risk of relapses in cHL patients. For instance, it has been shown that gains of chromosome 16p on HRS cells are associated with inferior outcome. 148 Interestingly, the expression of ABCC1, an oncogene located in the consensus region associated with gains of chromosome 16p, is positively correlated with refractory disease and risk of treatment failure in cHL patients. 148 Copy number amplification of more genes manifested as higher expression (eg, CSF1R (5q34), IL-10 (1q32)) or gain of function mutations (eg, STAT6 (12q13)) has also been implicated in HL. 149
Conclusions
The emergence of novel detection techniques has allowed major advances in identification and characterization of human chromosome variation, including CNAs, for their clinical potential in cancer. While karyotyping and FISH are still the most commonly used methodologies in the detection of CNAs or CNA-related genomic aberrations, NGS technologies have gained increasing attention as an important tool in both the discovery of novel CNAs and the daily practice of diagnosis in lymphoma. CNAs or CNA-related genomic aberrations have become an important index for diagnosis, risk stratification, and therapeutic guidance of lymphoma. However, due to the high heterogeneity in both disease entities and variable forms of CNAs in lymphoma, much more studies are required to discover, characterize, and translate clinical-relevant CNAs, which should then being comprehensively integrated with established genetic features, including SNVs, InDels, and fusion genes to achieve better disease management and therapeutic outcome.
Footnotes
Acknowledgments
Not applicable.
Ethical Considerations
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Author Contributions
XZ: Writing - Original Draft; ZY: Writing - Original Draft; SY: Drafted figures and tables; MZ: Reference collection; ST: Conceived the work; ZZ: Writing-Editing, Supervision; YL: Writing-Editing; WR: Writing-Editing. All authors contributed to the article and approved the submitted version.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability
Not applicable.
Artificial Intelligence (AI) Author Disclosure Statement
The authors certify that no AI tools were used in the creation of text, references, images, or any other content included in this manuscript. Furthermore, no scientific data has been generated or modified using AI. The authors take full responsibility for the accuracy, originality, and integrity of all submitted material.