
Abstract
Purpose
Hepatocellular carcinoma (HCC) is the most common liver malignancy in the world, and tumor-infiltrating T cells have been shown to be closely related to the prognosis of HCC. This study investigated the potential and efficacy of T cell receptor (TCR) repertoire characterization as a biomarker for predicting survival differences.
Methods
In this study, we used high-throughput sequencing technology to systematically analyze the characteristics of TCR repertoires in tumor tissues obtained from 23 long-survivors and 8 short-survivors diagnosed with HCC.
Results
The TCR composition in HCC long-survivors was found to be less diverse than in the short-survivors. In addition, in the context of V and J gene segments, long-survivors showed significantly higher usage of TRBJ1-3, TRBV10-1, TRBV15, and TRBV6-5, and lower usage of TRBJ2-2. Both principal component analysis (PCA) and the motif diagram of complementary determination region 3 (CDR3) sequences could clearly discriminate short- and long-survivors. And there were five up-regulated and one down-regulated CDR3 sequences in the LS group compared with the SS group. According to the characteristics of TCR repertoire, we also established the survival-related evaluation system and the prediction model for the survival period of HCC patients.
Conclusion
Our study adds to the existing knowledge of TCR rearrangement profiles in HCC patients by elucidating the differential TCR rearrangement profiles between long-term and short-term surviving HCC patients. Also, our analysis identified a number of TCR genes that are significantly associated with survival, and these may not only serve as prognostic biomarkers but may also play an important role in antigen-specific immunotherapy.
Keywords
Introduction
Primary liver cancer has been one of the most common cancers in the world, of which hepatocellular carcinoma (HCC) accounts for 85%-90%. 1 As the third leading cause of cancer-related mortality, HCC leads to over 800,000 deaths annually, with an average survival period of 6 to 12 months. Moreover, 80% to 90% of HCC cases are secondary to chronic liver diseases, such as hepatitis or cirrhosis.2,3
Currently, liver transplantation, surgical resection, and radiofrequency ablation are the primary curative treatments for HCC. 4 Unfortunately, the 5-year postoperative recurrence rate was 30% after liver transplantation and 70% after hepatic resection, 5 which severely hampers the long-term survival of HCC patients. 6 There have been many achievements in the field of HCC research, such as the use of ATG-4B mRNA as an independent prognostic factor in the prognosis and diagnosis of HCC with important clinical significance. 7 At the same time, there are many studies exploring the characteristics and potential of TCR in HCC patients. Current studies have found no significant differences between the TCR diversity of tumors and matched adjacent normal tissues in HCC patients, but there are some differences in the expression between gene segments on the TCR chain that may serve as biomarkers for the diagnosis and treatment of HCC.8,9 However, current treatment regimens have high relapse rates, cannot accurately predict the time to relapse or the situation before relapse and intervene in treatment, and lack biomarkers to predict disease progression and prognosis.
Tumor-infiltrating lymphocytes (TIL) play a pivotal role in the occurrence and outcome of various tumor types, which are selectively activated and expanded via specific recognition of tumor neoantigens. 10 The killing effect of TIL was found to be associated with the efficacy of immunotherapy, chemotherapy, and radiotherapy.11,12 Thus, an in-depth analysis of T cells may help accurately understand the individual tumor immunity in HCC. 13 The complex clonal composition of TIL is determined by somatic recombination of the T cell receptor (TCR) α- and β-chains, which can be investigated by high-throughput sequencing (HTS). 14 Recently, a number of studies have suggested that TCRβ characteristics might be used as a good biomarker for early diagnosis and prediction of prognosis in HCC. For instance, Han et al investigated the TCR repertoire by sequencing and found the significantly different, highly expanded clone (HEC) ratio between liver cancer patients and healthy controls. 15 Chen et al also performed TCR sequencing and observed higher TCR diversity in HCC biopsies compared with adjacent non-tumor tissues. 16 While several studies reported that some HCC patients tend to live longer after their diagnosis than others,17,18 the detailed connection between TCR repertoire and long survival in HCC remains unclear. T cells recognize antigens through T-cell receptors (TCRs) expressed on the cell membrane. The diversity of TCR repertoire is mainly determined by complementary determination region 3 (CDR3) generated by genomic rearrangement of the variable (V), diversity (D), and joining (J) regions. 13
High-throughput sequencing (HTS), with its high sensitivity and high throughput, provides detailed data to support the unraveling of complex immune microenvironments. With the ability to identify multiple immune types simultaneously, high-throughput sequencing can reveal complex molecular properties that are difficult to detect by traditional methods. However, its technical variability may lead to limited reproducibility and consistency of results, affecting the interpretation of data; meanwhile, high-throughput sequencing faces the challenge of high cost in clinical applications, which may limit its popularization in routine diagnosis and treatment. Therefore, high-throughput sequencing of the CDR3 region on the TCR could facilitate our understanding of the adaptive immune state of the organism. Recently, several studies have illuminated that CDR3 diversity of TCR can serve as a relatively stable biomarker, contributing to early disease diagnosis, efficacy monitoring, and prognostic evaluation.19,20
In the present study, we performed high-throughput TCR sequencing and compared the characteristics of tumor-infiltrating T cells between long- and short-survivors diagnosed with HCC, so as to uncover public TCR response supporting longer survival. The results we present could provide a better understanding about infiltrating T lymphocytes in liver cancer and allow the improved stratification of the HCC patients for treatment selection and prognosis prediction.
Materials and Methods
Patients and Samples
The reporting of this study conforms to REMARK guidelines. 21 HCC patients with clinical and survival information were enrolled at hospital. None of the patients had received chemotherapy or radiotherapy or had other immune-related diseases. This study included patients with a diagnosis of primary hepatocellular carcinoma (HCC) confirmed by imaging or pathology, aged 18-70 years, of either sex, with an ECOG score of 0 or 1, a Child-Pugh score of A for liver function, and who were not receiving any form of anticancer therapy or had metastatic disease; no serious abnormalities in laboratory tests, and with normal or mildly abnormal renal function, hematology, and infection markers, who signed an informed consent. Exclusion criteria included previous history of other malignant tumors, chronic immune system disease, uncontrolled HBV or HCV infection, serious complications (such as cardiovascular disease, hepatic encephalopathy or ascites), recent use of immunosuppressant drugs or vaccination, pregnancy or lactation, and psychiatric or cognitive disorders to ensure that there were no confounding factors in the study population. All participants provided written informed consent before enrollment. All patients were classified into two groups based on survival time: (1) short-survivors (ie, the SS group), who succumbed to the disease within 12 months from diagnosis, and (2) long-survivors (ie, the LS group), who survived for at least 24 months from diagnosis. Patients with survival time in the range of 12 to 24 months were not included in this study. This study was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee. The tumor samples were collected immediately after surgical resection and washed with ice-cold saline, then immersed in liquid nitrogen for further analysis.
Laboratory Examination
Routine laboratory examinations were performed as follows: Liver/kidney function and electrolytes were evaluated by using the fully automatic biochemistry analyzer Cobas (Roche Diagnostics, Germany), blood routine examination was conducted by using the fully automated hematology analyzer XN-10 (B4) (Sysmex, Japan), Coagulation was measured with the fully automated coagulation analyzer CS5100+ (Sysmex, Japan), Tumor markers were evaluated by using the fully automatic chemiluminescence immunoassay pipeline A3600 + i2000SR (Abbott, America), according to the manufacturer's instructions.
TCR Repertoire Sequencing
Total RNA was extracted from tissue samples using the RNA simple Total RNA Kit (Tiangen Biotech, Beijing, China). The concentrations of RNA were evaluated using a NanoDrop ND-2000 spectrophotometer (Thermo Scientific, UK). cDNA synthesis and multiplex PCR amplification of all possible rearranged TCR β-chain sequences were performed together using the Immune Repertoire Library Preparation Kit (Geneway, Jinan, China) following a protocol described in a previous study. 22 TCR repertoires were sequenced on the DNBSEQ-T7 platform (MGI, Shenzhen, China), generating paired-end short reads with 150 bp in length. Based on the sequence characteristics of TCR, TCR-related sequences in total RNA were specifically extracted to be screened, enriched, amplified, and ultimately built into libraries for sequencing.
Sequencing Data Analysis
The sequencing data stored in FASTQ format were primarily processed to discard the low-quality sequences, with the remainders reserved for subsequent analysis. We filtered reads that did not meet the following criteria: (1) reads with an average reads quality of less than 20; (2) reads with a greater than 20% share of bases with a base quality of less than 10; (3) reads with a greater than 5% share of N; and (4) reads that contained sequencing adapter sequences. The reference TCR sequences of V and J genes were downloaded from the International ImMunoGeneTics (IMGT) database (http://www.imgt.org/vquest/refseqh.html). 23 The MiXCR software 24 was used to map the sequencing data to the reference sequence. The frequency of V and J genes was converted into rpm (reads per million) for standardization. The diversity of TCRβ was measured by Shannon and Simpson indexes, which have been widely used for assessing the TCR characteristics as previously described. 25 A circular diagram was created using the Circos software 26 to visualize the V-J gene pairs.
Statistical Analysis
T-tests and Fisher's exact tests were performed using R software (version 4.0.2) to identify the differences between the SS and LS groups in terms of demographic and clinical variables, with statistical significance set at P < .05. A two-sided P-value <.05 was considered statistically significant. Principal component analysis (PCA) was used for clustering analysis. Principal Component Analysis (PCA) is a statistical method designed to downscale high-dimensional data through linear transformations while retaining the main information in the data. PCA has significant advantages in reducing dimensionality, reducing redundant features and revealing underlying data structures. In this study, a survival difference classifier was constructed by the Random Forest (Random Forest) algorithm for predicting the survival outcomes of hepatocellular carcinoma patients. During the model training process, cross-validation techniques are used to evaluate and optimize the performance of the classifiers, alternating between model training and testing to reduce the bias due to differences in data distribution and improve the reliability and stability of the model prediction results.
Results
Patient Characteristics
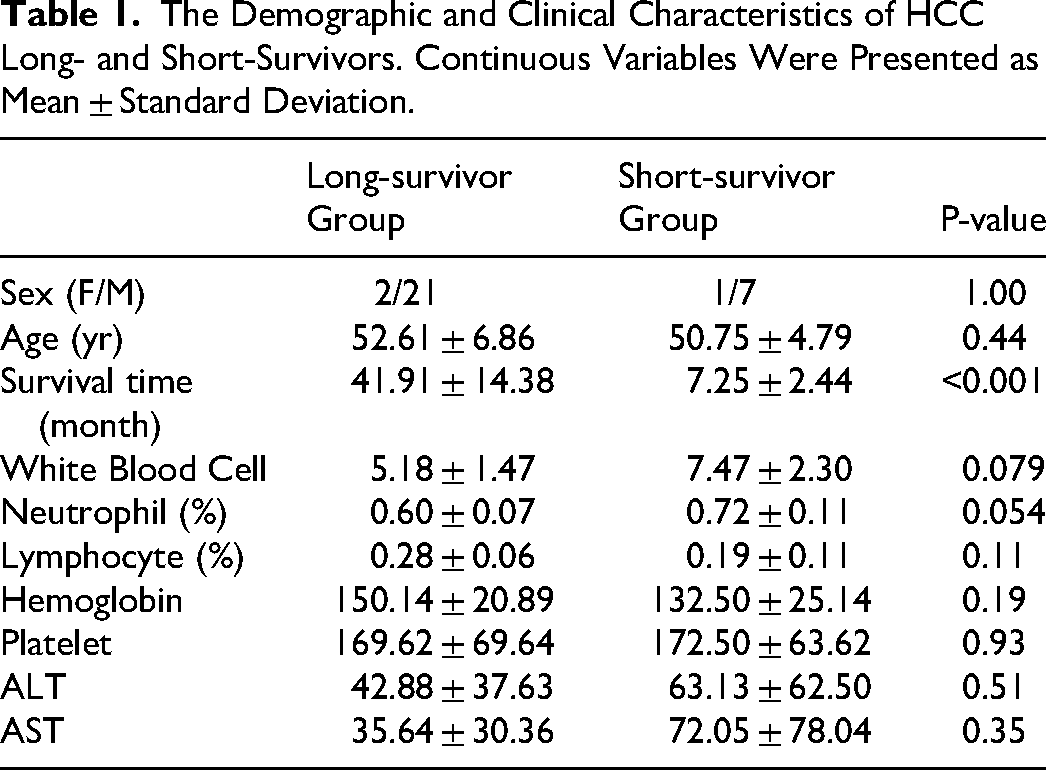
A total of 31 HCC patients were diagnosed after surgical resections and enrolled in this study, including 23 long-survivors (LS) and 8 short-survivors (SS). A summary of clinicopathological characteristics was presented in Table 1. Except for survival time, two groups showed no significant difference in demographic and clinical variables, including sex, age, immune parameters, and liver function indexes.
The Demographic and Clinical Characteristics of HCC Long- and Short-Survivors. Continuous Variables Were Presented as Mean ± Standard Deviation.
Quality of Sequencing Data
We performed multiplex PCR to amplify the TCRβ genes and conducted high-throughput sequencing to examine the clonotypes and V/J gene segments (see Materials and Methods). The sequence profiles of all HCC patients were shown in Table S1. After the removal of low-quality reads, a total of 456.6 M productive TCRβ sequences were collected from SS and LS samples, with an average of 14.7 M per sample. Except for the average number of unique J gene segments in LS samples being slightly lower than that in SS samples (t-test, P = .04), LS and SS groups had no significant difference in various quality control indexes, including clean reads number, average read length, V gene count, and the number of unique V-J combinations (Figure 1).
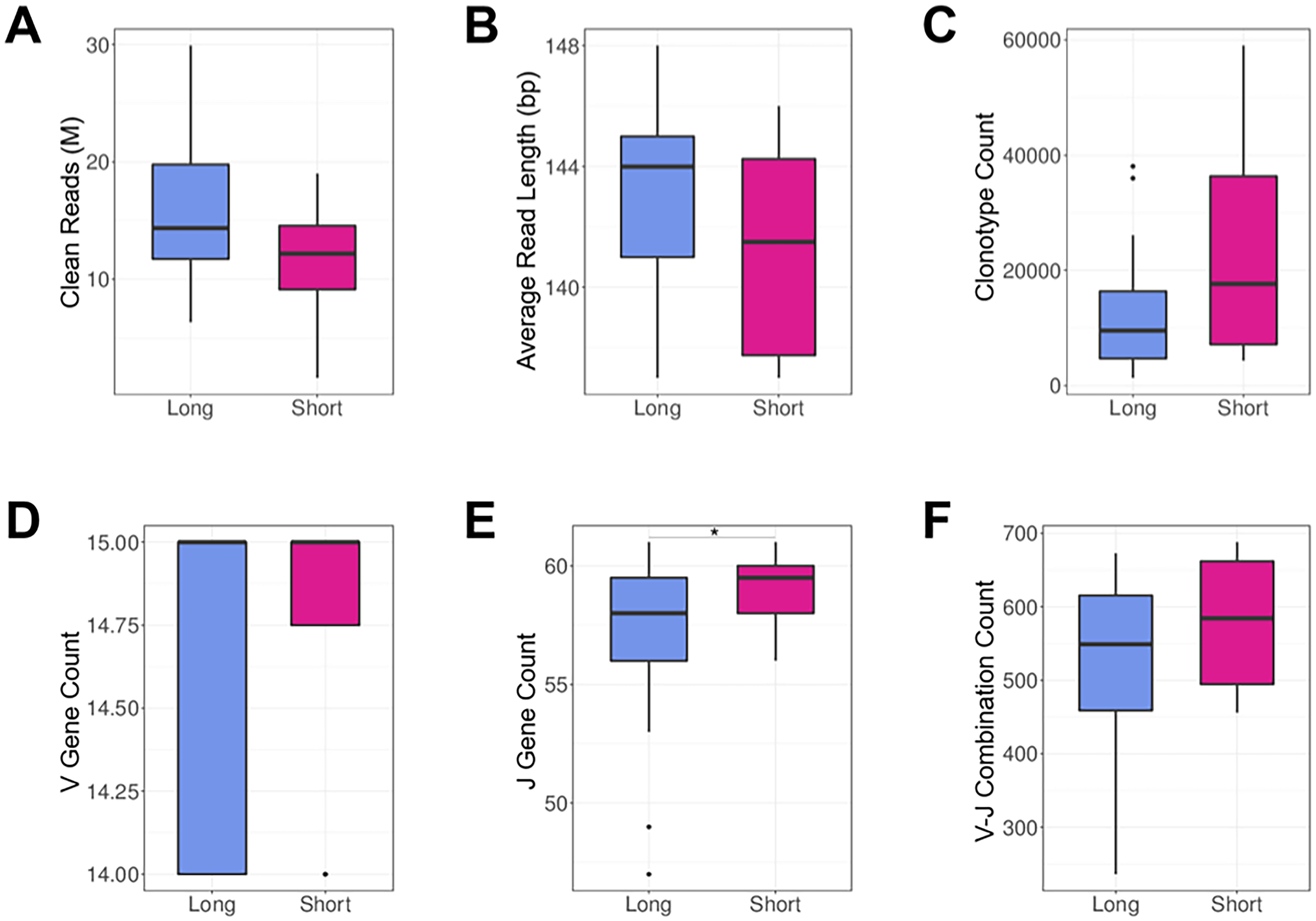
TCR sequencing metrics in LS and SS groups. (A) Clean reads. (B) Average read length. (C) Number of unique clonotypes. (D) Number of unique V genes. (E) Number of unique J genes. (F) Number of unique V-J combinations. Boxplots displayed the distribution of data with minimum, first quartile, median, third quartile, and maximum values.
TCRβ Diversity of LS and SS Patients
TCR expression profiles in the tumor samples from long- and short-survivors were subsequently analyzed to assess their systemic immune responses. We calculated the Shannon and Simpson indexes to estimate the diversity of TCR clonotypes in each sample, which were not relevant to the variation of sample sequencing depth. Both Shannon and Simpson indexes were significantly higher in the tumor tissue of SS subjects than in LS groups (t-test, P < .05, Figure 2A and B), suggesting that the TCR composition in HCC long-survivors was more concentrated than in the long-survivors. Such differences were further illustrated by treemap plots, with each rectangle in the plot representing a unique clonotype and the size of the rectangle denoting the relative frequency in the TCR repertoire (Figure 2C and D). It was observed that certain TCR clonotypes were prone to clonal expansion in LS individuals.

The comparison of the TCR repertoire diversity indexes between LS and SS groups. (A) Shannon index. (B) Simpson index. (C) Treemap of TCR clones in a typical long-survivor example. (D) Treemap of TCR clones in a typical short-survivor example.
Principal Component Analysis (PCA)Between LS and SS Groups
We further performed Principal Component Analysis (PCA) on the V-J combination frequency profile. As shown in the PCA plot (Figure 3), a difference was found between LS and SS groups. The LS group was looser than the SS group and indicated that there was a broader spectrum of immune defense characteristics in the LS group.

PCA in the LS and SS patients. PCA results showed that LS group was distinguished from SS group. PC1 indicates the first principal component, whereas PC2 refers to the second principal component.
Differential Usage of V and J Gene Segments
A total of 59 V and 13 J gene segments were identified in the LS and SS groups. The top four frequent V gene segments were TRBV19, TRBV20-1, TRBV23-1, and TRBV27 (Figure 4A). The top four frequent Jβ segments were TRBJ1-1, TRBJ1-2, TRBJ2-2, and TRBJ2-7 (Figure 4B). At the same time, compared with the SS group, the LS group showed significantly higher percentages in the clonotypes of TRBJ1-3, TRBV10-1, TRBV15, and TRBV6-5, and a lower percentage in TRBJ2-2 (Figire 4C and D). These gene differences indicate to us the characterization of the LS and SS groups in the direction of TCR genes.
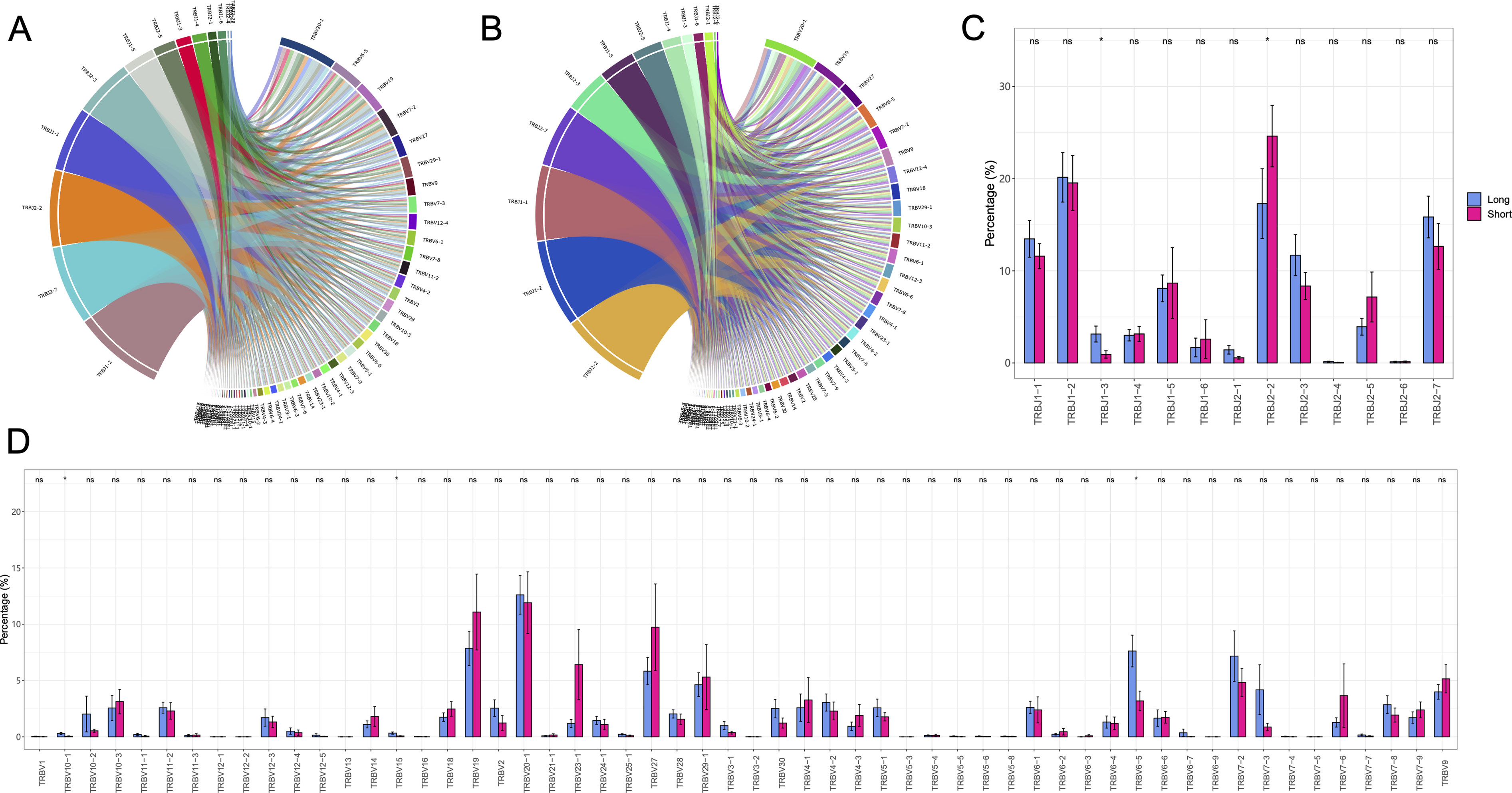
Differential usage of V and J gene segments between LS and SS individuals. (A) Circular diagram of V-J gene combinations in LS group. (B) Circular diagram of V-J gene combinations in SS group. (C) J gene usage. (D) V gene usage. * P < .05.
Different CDR3 Sequences Between LS and SS Patients
We further analyzed and compared the abundance of CDR3 sequences between LS and SS groups. The volcano map showed that there were five upregulated and one downregulated sequences between the LS and SS groups (Figure 5A). And the scatter diagram showed the 6 different expression sequences in LS and SS groups (Figure 5B). Also, the thermal map showed the differentially expressed amino acid in each sample of the LS and SS groups (Figure 5C), which had the potential as the diagnostic marker of LS and SS HCC patients.
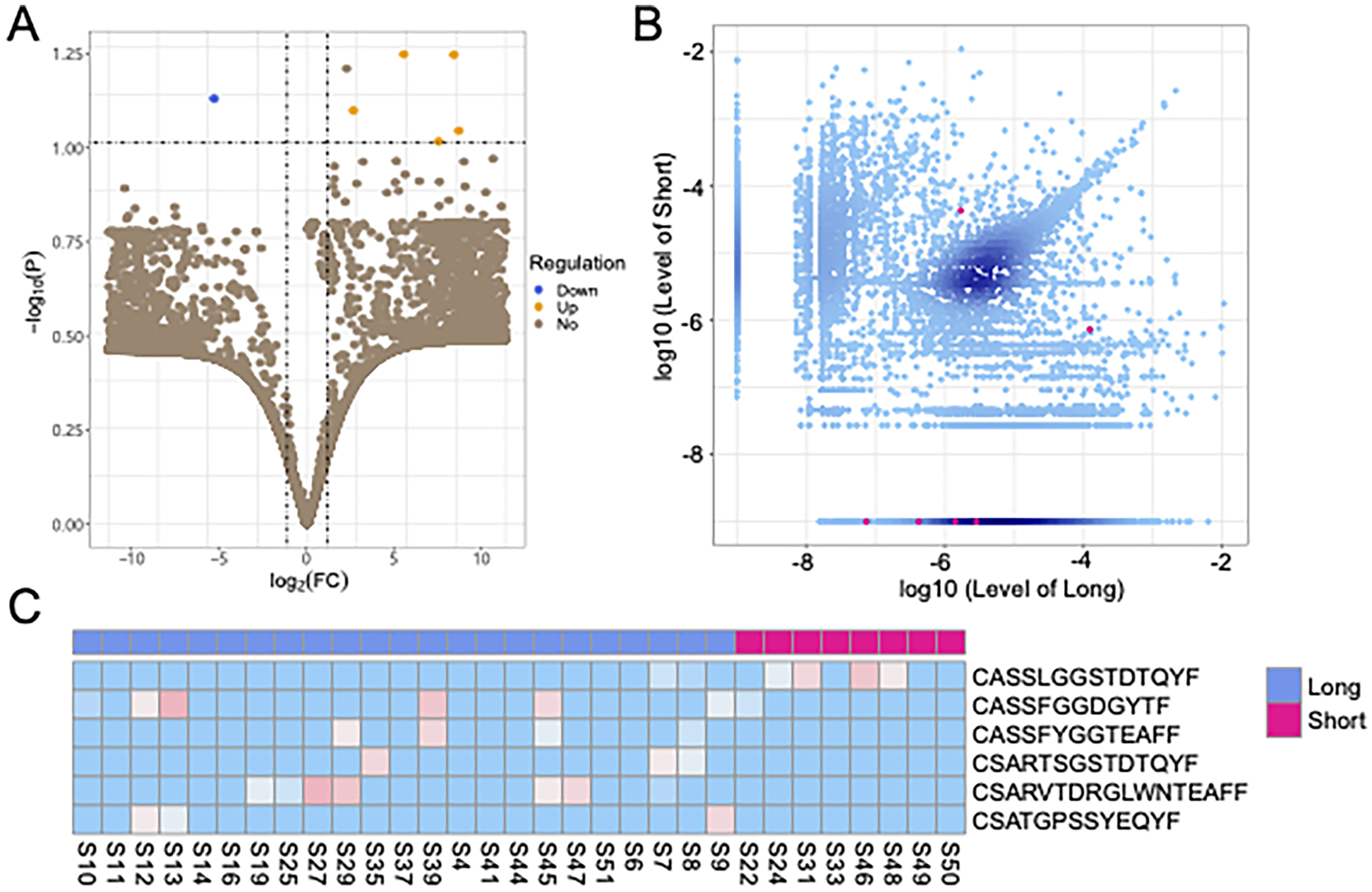
Different expression of CDR3 sequences between LS and SS patients. The volcano map(A), the scatter diagram (B), and thermal map (C) showed the amino acid sequences of differential expression in LS and SS groups.
The Motif Specificity of TCR Repertoire and Survival-Related Evaluation System
We aligned the alignment of the top 50 abundant CDR3 sequences with the indicated length to make a motif diagram. The results showed that the motif was different between the LS and SS groups (Figure 6A and B), which suggested that selecting a suitable amino acid sequence can effectively distinguish LS and SS patients of HCC. Based on the different characteristics of TCR repertoires between the LS and SS groups, we designed a set of survival-related evaluation systems, containing the abundance information of specific CDR3 sequences, diversity indicators, and so on. We used this evaluation system to score the 31 samples and conducted the overall survival analysis, combining the clinical information of the patients. We found that the evaluation system could effectively distinguish HCC patients with different survival periods (P = .014, Figure 6C).
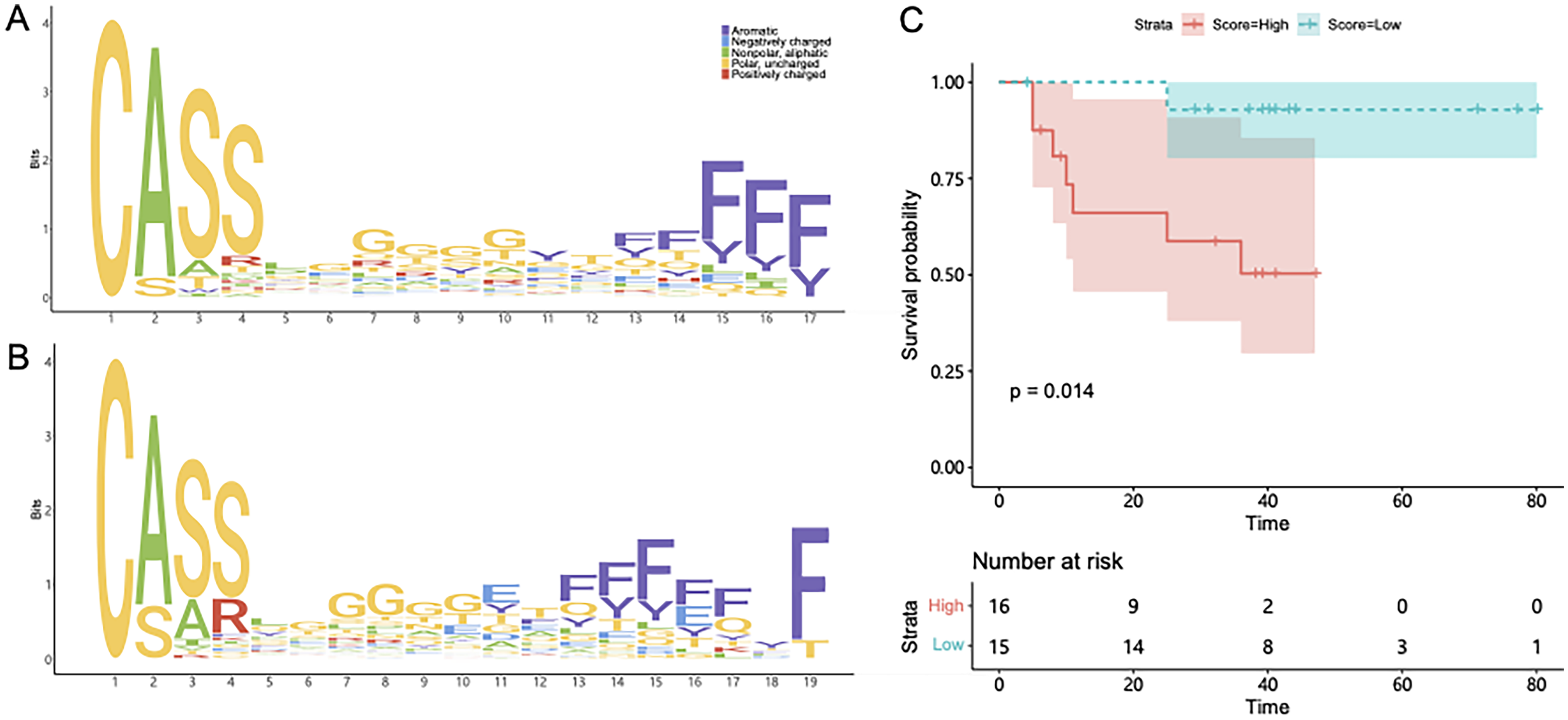
The motif specificity of TCR Repertoire and survival-related evaluation system. (A) The motif diagram of top 50 abundant CDR3 sequences in LS group. (B) The motif diagram of top 50 abundant CDR3 sequences in SS group. (C) Survival curves of LS and SS group from our survival-related evaluation system. Red indicates samples with high scores and blue indicates samples with low scores.
The Prediction Model for the Survival Period of HCC Patients
Based on the different characteristics of TCR repertoires between the LS and SS groups, we developed a prediction model for the survival period of HCC patients using the random forest algorithm. In order to make the model accurate, we adjusted the parameters from 0.2 to 0.3, which minimized the fault tolerance rate (Figure 7A). The result suggested that the model had a stable function for classification. Then, we evaluated the model classification effect for the survival period of HCC patients. The results showed that the model could make a clear distinction between LS and SS patients (AUC = 0.456-0.849), and the distribution of the ROC curve was relatively smooth (Figure 7B). Moreover, we measured the V-J combinations that influenced the model evaluation effect and found that the top ten combinations contributed the most to the model (Figure 7C).

The random forest prediction model for the survival period of HCC patients. (A) The parameter selection for this model showed the error rate for the classification of LS and SS groups under different tree number. (B) ROC curve showed the classification effect of the model. The ordinate was true positive rate (sensitivity), and the abscissa is the false positive rate (1-specificity). (C) The top 10 V-J combinations that affected the model effect. Mean Decrease Gini is a rating index, and its value is positively correlated with the degree of effect on the model.
Discussion
Hepatocellular carcinoma (HCC) is one of the most prevalent causes of cancer, with a generally high mortality rate and poor prognosis.27,28 Previous research has proved that effective biomarkers could identify HCC patients with longer survival times. 29 Evidence suggests that HCC malignant cells can induce a strong immune response, which creates the proper microenvironment for their growth. 30 Since the TCR repertoire mirrors the status of the immune system, 15 it is necessary to analyze TCR diversity to depict the immunosurveillance and its association with HCC prognosis.
The researchers previously reported a characterization of the HCC immune contexture and describe an immune-specific class using a non-negative matrix factorization algorithm. 31 And they also performed RNA and whole-exome sequencing, TCR-sequencing, multiplex immunofluorescence and immunohistochemistry in a novel cohort of 240 HCC patients and validated the results in other cohorts comprising 660 patients. Therefore, they characterized the immunogenomic contexture of HCC and defined inflamed and non-inflamed tumors, which could help predict immune response in HCC. 32 However, the immunological characteristics of HCC patients with different survival periods have not been reported yet.
In this study, we comprehensively investigated the TCR repertoires of long- and short-survivors of HCC by using the powerful TCR sequencing technology. Compared with traditional methods, such as quantitative PCR and flow cytometry assays, TCR repertoire analysis by high-throughput sequencing enables generation of an extensive snapshot of the immune system.33,34 While a number of factors may contribute to the overall survival of HCC patients, we specifically explored the T cell immune responses that are associated with survival differences in HCC patients with similar clinical characteristics.
The profound relationship between TCR diversity and HCC prognosis has yet to be fully understood. Although significantly higher TCR diversity was observed in tumor biopsies compared with adjacent non-tumor tissues, 16 a previous study suggested no correlation between TNM stage or prognosis of HCC and intra-tumoral TCR repertoire diversity. 9 We found that the TCR diversity is significantly higher in the SS group than that in the LS group. Some research reported similar results that a less diverse TCR repertoire in tumor tissue is associated with improved overall survival of non-small cell lung cancer. 35 We have provided further evidence that a more focused TCR repertoire might mark a stronger T cell response to tumor-related antigens, which could benefit better cancer prognosis.
Immunotherapy is a promising method for treating advanced PC, and remarkable progress has been made in adoptive T-cell therapy and so on. T cells can stimulate tumor-infiltrating lymphocytes (TIL) to activate the adaptive immune system and infiltrate a large number of solid tumors, thereby killing tumor cells. Evidence suggests that immune activation is expected to increase the survival rate and prolong the life expectancy of cancer patients. 36 Hence, immunotherapy is regarded as a promising approach for treating HCC,35,37 but limited efficacy of immunotherapy in a subpopulation of patients remains a crucial issue.38,39 In our analysis of TCR sequencing data, we found that there were some of the V and J gene segments (ie, TRBJ1-3, TRBV10-1, TRBV15, and TRBV6-5) were significantly over-expressed in long-survivors, and 5 up-regulated and 1 down-regulated CDR3 sequences, indicating that intra-tumoral T cell immune responses to some specific antigen targets might benefit the prognosis. Therefore, developing treatments to target these antigens or isolating antigen-specific TILs could effectively aid the patients non-responsive to regular immunotherapy. According to the characteristics of TCR repertoire in HCC patients, we established the survival-related evaluation system and survival period prediction model and verified its reliability, which indicated that HCC patients with different survival can be clearly distinguished by TCR repertoire characteristics.
Changes in TCR repertoire are closely related to the immune response in HCC patients and may have important clinical significance for the prognosis of HCC patients. The diversity of TCR profiles in HCC patients is usually low, and the amplification of specific TCR clones is closely related to tumorigenesis, progression, and metastasis. By analyzing TCR β-chain sequences in peripheral blood or tumor tissues of HCC patients, the key role played by specific TCR clonotypes in immune escape and drug resistance of malignant tumor cells can be uncovered, making TCR profiles a potential prognostic marker. Meanwhile, changes in the diversity of TCR profiles and assessment of clonal amplification patterns can provide valuable information about immune tolerance and immune escape in patients. Combining TCR profile analysis with other clinical indicators (eg, tumor stage, liver function, etc) can provide a personalized prognostic assessment of HCC patients and help predict treatment response. Therefore, TCR repertoire is not only a marker of immune escape in hepatocellular carcinoma but also an important tool for predicting patients’ prognosis in clinical practice. In the future, the in-depth study of TCR profiles by high-throughput sequencing technology, combined with the molecular features and immune microenvironment of hepatocellular carcinoma, may provide new ideas and directions for early diagnosis, precise treatment, and prognosis judgment of HCC.
By sequencing the TCR sequence, researchers were able to analyze the T-cell clonal populations of HCC patients and assess the strength and diversity of anti-tumor immune responses. This offers the potential for individualization of immunotherapy. In immunotherapy strategies in general, and immune checkpoint inhibitors (eg, PD-1/PD-L1 antibodies) and T-cell therapies (eg, CAR-T therapies) in particular, the analysis of TCR sequences can help to determine which T-cell clones have specificity against tumors, thereby improving the targeting and efficacy of the therapy. In addition, TCR sequencing can reveal immune escape mechanisms in the immune microenvironment of HCC, such as how tumors evade immune surveillance by altering the antigens recognized by the TCR. Therefore, combining the analysis of TCR sequences with immunotherapy strategies can optimize treatment regimens, identify potential immune escape mechanisms, and provide more effective individualized treatment for patients.
There are also some key factors in this study that limit our results. First, our sample size is not large, especially for short survival periods. Subsequently, we will continue our research in this direction and keep increasing the number of experimental samples to obtain sufficient statistical utility and validate the current results. Second, due to technical limitations, we did not construct T cell subsets in this study, and we will consider sequencing T cell subsets (eg, CD4 + and CD8+) separately by flow cytometry in order to obtain more accurate and reliable results. 40
Conclusion
In summary, we characterized T-cell receptor repertoires of long- and short-survivors with HCC by high-throughput sequencing. Our findings demonstrated that the TCR diversity and V/J gene usage of short-survivors were distinct from those of long- survivors. And differential expression of specific V, J genes and CDR3 sequences exhibited association with longer survival in HCC. It suggested that there may exist novel antigens in HCC tumors, which could be used to design prognostic biomarkers and targeting immunotherapies in the near future.
Supplemental Material
sj-docx-1-tct-10.1177_15330338251329699 - Supplemental material for Characterization of T-Cell Receptor Profiles Predicts Survival Situation in Patients with Hepatocellular Carcinoma
Supplemental material, sj-docx-1-tct-10.1177_15330338251329699 for Characterization of T-Cell Receptor Profiles Predicts Survival Situation in Patients with Hepatocellular Carcinoma by Fengyan Wang, MD, Li Deng, MD, Ziqiang Li, MD, Qiwei Cao, MD, Runze Jiang, PhD, Changqing Xu, MD and Jing Yang, MD in Technology in Cancer Research & Treatment
Footnotes
Acknowledgements
The authors wish to thank all the patients participating in the study and all the caregivers and physicians in the First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital.
Ethics Approval and Consent to Participate
This study was approved by the Ethics Committee of the First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital. Informed consent was obtained from each patient for their participation.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the Shandong Provincial Integrated Traditional Chinese and Western Medicine Special Disease Prevention Project (No. YXH2019ZXY003), the Technology Research and Development Project of Shandong Province (Public Welfare Project) (No. 2019GSF108167), the National Natural Science Foundation of China (No. 81200275), the Natural Science Foundation of Shandong Province (No. ZR2012HL20), and the Medical and Health Technology Development Program of Shandong Province (No. 2015WS0221).
Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.