
Abstract
Introduction
Uterine carcinosarcoma (UCS) is a rare, aggressive and lethal gynecological cancer in women of all ages that accounts for 3%-7% of all uterine cancers, with a higher incidence in women aged 50-70. 1 Uterine carcinosarcoma include leiomyosarcoma, carcinosarcoma, and endometrial stromal sarcoma according to common histological types. The prognosis of patients with uterine carcinosarcoma is usually poor due to its aggressive and early metastatic nature. A recent US Surveillance reported a 42% five-year overall survival (OS) rate for uterine leiomyosarcomas patients, and all patients with late stage (stage III-IV) uterine leiomyosarcomas dead within 5 years. 2 Most patients have advanced-stage disease at presentation, and the majority of patients with US have a dismal prognosis; surgical resection is the preferred treatment option for patients with early-stage disease, because available systemic therapies are of limited effectiveness. Therefore, an increased understanding of its molecular tumor biology is urgently needed. In recent years, some studies, such as L1CAM have a prognostic role in stage I endometrial cancer, 3 with some clinical studies about the role of the excised vaginal cuff length as a prognostic factor in terms of DFS and recurrence rate/site in low-risk endometrial cancer patients, 4 has enhanced and expanded our horizons in the discovery and treatment of gynecologic oncology.
The mechanism behind UCS development is unresolved. Current hypotheses include collision, combination, and conversion mechanisms. The current consensus favours the conversion theory. This occurs via epithelial-mesenchymal transition (EMT), wherein epithelial cells transform into migratory mesenchymal cells. Supporting evidence includes immunohistochemical and molecular evaluations demonstrating co-expression of epithelial markers in both cancer and sarcoma constituents, along with similarities in genetic alterations.5,6 The symptoms of UCS are similar to other uterine adenocarcinomas, such as bleeding, abdominal pain and uterine enlargement. 7 Pathological sampling is difficult to confirm UCS, and there is insufficient data to show that CT and MRI are diagnostically useful or cost effective before surgery. 8 The findings of elevated preoperative tumor markers (such as CA125, PAX8, and PCNA) were not confirmed by an affirmative study yet.9,10 Due to UCS's low incidence and few samples, much of the data on UCS treatment is retrospective rather than prospective. Therapies for UCS include surgery (for early-stage and advanced-stage), chemotherapy, radiotherapy and combination therapy. For the chemotherapy of UCS, HER2 is a therapeutic target. One of the HER2-targeted therapies, Trastuzumab Deruxtecan (T-DXd), has been recently proved to be effective in both HER2-high and HER2-low advanced patients. 2 Diagnosis of developing UCS is potentially based on RNAs, which participate in a variety of biological processes. Therefore, our study explored the biological role of RNAs in developing UCS through whole transcriptome sequencing.
In recent years, the role of ncRNAs in oncology has been greatly recognized. Previously, ncRNAs were thought to be non-functional that could not be translated into proteins, but subsequent studies have revealed that ncRNAs may be involved in biological behaviors such as growth, development, and organ function, especially in tumors. A recent study shows ncRNA derived peptides are modulators of tumor behaviors. 11 A shocking study reveals genome is pervasively transcribed into RNAs, but they are seemingly not translated into proteins. 12 Some research show miRNAs are possible biomarkers in oncology. Pathway activation on miRNA expression patterns involve in and influence targeted therapy for tumors. 13 The role of circRNA has also been elucidated as an anti-tumor immune and biomarker.14,15 The above researchs show that ncRNA is a novel and important breakthrough in oncology research. In this study, whole-transcriptome sequencing was performed based on the UCS and their corresponding paracancerous tissues. Subsequently, RNAs expression analysis was performed of the UCS and Normal groups. We construct a miRNA-circRNA-gene co-expression network to provide possible breakthroughs for biological behaviors and potential markers for diagnosis and treatment in developing UCS.
Materials and Methods
Patients and Tissue Samples
Five patients with uterine cancerarcoma who were consulted and surgically resected in the Department of Gynaecology, Suining Central Hospital in 2019-2021 were collected. Specific information about this cohort study can be found on the STROBE checklist in Supplemental Material. All sample were collected with the informed consent of the patients. The cancer tissues and matched paracancerous tissues of the three cases were collected as controls. Clinical and pathological characteristics were also collected for each patient (Table 1). Tissue samples were collected during surgery (resection) and kept formalin fixed paraffin embedded (FFPE) blocks. One part of the samples were processed for the whole-transcriptome sequencing, and the remained parts were used for pathological and histological assessments. This research is approved by Ethics Committee of Zunyi Medical University Affiliated Hospital. Ethics approval number is Zun Yi Lun Shen (2024) 1-028.
Clinical and Pathological Characteristics of 5 UCS Patients.

The header row list patients ID, tumor type, OS (day), death, PFS (day), tumor size, postoperative stage and location.
Libr4.2 Library Construction and Transcriptome Sequencing
Utilizing the Omicshare Cloud Platform (Guangzhou, China), eukaryotic mRNA was enriched with a polyA tail via magnetic beads bearing Oligo(dT). Sonication shattered the mRNA, serving as a template for cDNA synthesis using random oligonucleotides as primers, with M-MuLV reverse transcriptase. RNA strands were destroyed with RNaseH and a double-stranded cDNA synthesized with dNTPs catalyzed by DNA polymerase I. The double-stranded cDNA was end-repaired, A-tailed and coupled to the sequencing junction, then subjected to AMPure XP (Beckman Coulter) bead screening, PCR amplification, and purification to generate the library. These libraries were sequenced on the Illumina HiSeqTM Platform. RNA purity and integrity were evaluated using the NanoPhotometer spectrophotometer and Agilent 2100 Bioanalyzer (Agilent Technologies). Quantification of all RNA was performed on the Qubit2.0 Fluorometer (Thermo Fisher Scientific).
Analysis Process
Upon acquiring the downstream data, it is initially refined to yield HQ Clean Reads. These HQ Clean Reads of each sample are contrasted against the GENCODE V36 (GRCh 38/hg38) reference genome separately to generate individual comparison outcomes.
Differentially Expressed lncRNA Analysis
lncRNA differential expression analysis utilizes DESeq2-derived readcount data for lncRNA expression profiling. EdgeR is used for non-replicated or sample-to-sample variation studies. The analysis involves readcount normalization, pvalue formation, and FDR determination with multifaceted test correction. A volcano map displays significant lncRNA variations between comparison groups, with lncRNAs closer to the extremes indicating higher disparities. Hierarchical clustering of differential lncRNA expression patterns, followed by heat map representation, aids in identifying functional correlations. Each lncRNA undergoes z-score computation before visualization.
CircRNA Identification and Quantification
RNA extracted from samples is initially treated to eliminate ribosomal RNA. The resultant circular RNA is then random fragmented into 200-nt segments. This fragmented RNA is utilized as a template for reverse transcription to create double-stranded cDNA. Both ends of the cDNA are repaired, and a splice is added before fragment size selection via agarose gel electrophoresis for PCR amplification. HQ Clean Reads from each sample are individually compared against the reference genome to yield sample-specific results. Resulting Unmapped Reads are then isolated, their ends are truncated (default 20 bp), to form Anchored Reads, which are then compared with the genome for identification of circular RNAs using find_circ. HQ Clean Reads are further compared with the reference genome to derive each sample's results. The circBase database (http://www.circbase.org/) is then updated with annotated circRNAs, defined as pre-existing circRNAs, and unannotated circRNAs, defined as newly predicted circRNAs.
Differentially Expressed Genes Analysis
We apply the limma (linear models for microarray data) technique, an advanced differential expression screening methodology, utilizing the R software package limma (version 3.40.6). This approach involves performing log2 transformation on the expression spectrum dataset, implementing lmFit for multiple linear regression, and subsequently utilizing eBays to calculate moderated t-statistics, moderated F-statistic, and log-odds of differential expression via empirical Bayes moderation of the standard errors towards a uniform value. The end result is the identification of differentially significant genes.
Construction of miRNA-circRNA-Gene Co-Expression Network
In order to delve into and represent the correlations of RNA molecules and genes, an miRNA-circRNA-gene coexpression network was crafted. Genes showing a Pearson's correlation coefficient exceeding 0.99 were identified as significant. The bioinformatics tool Cytoscape was employed to construct a coexpression network involving these miRNAs, circRNAs, and genes with notable correlation. Network analysis utilized the degree centrality metric to assess gene significance.
Results
Identification of Differentially Expressed lncRNAs in UCS
A total of 2092 lncRNAs were differentially expressed between UCS and paracancerous tissues. Comparing to paracancerous tissues, UCS groups have 1632 upregulated and 460 downregulated lncRNAs (Figure 1a, b). Result suggests that in the UCS group, the upregulated lncRNAs may be the indicating biomarker or the pathways involved in developing UCS. The lncRNAs data is available in Supplemental Table 1.

(a) Volcano plot of lncRNA. The volcano plot shows the fold change against the p value for lncRNAs. Blue and red dots represent lncRNAs with significant change (P < .05, FC > 1.5). Red dots represent 1632 upregulated lncRNAs and blue dots represent the 460 downregulated lncRNAs comparing to paracancerous tissues. Volcano plot shows the statistically significant expressed lncRNAs. (b) Heatmap of lncRNA. Red and blue colors show the highest and lowest correlation.
Differentially Expressed Genes (DEGs) Analysis
Employing the limma (linear models for microarray data) methodology, we employ the R software package limma (version 3.40.6), an advanced differential expression screening system. This entails log2 transformation of the expression spectrum, followed by lmFit for multiple linear regression and subsequent eBays application to compute moderated t-statistics, moderated F-statistic, and log-odds of differential expression through empirical Bayes moderation of the standard errors. The outcome is the identification of significantly differentially expressed genes. CDADC1, CADM3-AS1, AL133482.1, TTC7A, and DANCR are the top five DEGs in UCS samples, as per their P.Value ranking.
The volcano plot (Figure 2a) illustrates the whole-transcriptome sequencing DEGs. The heatmap (Figure 2b) portrays the DEG and sample clusters. GSEA analysis (Figure 2c) displays twelve upregulated and downregulated pathways in the gene expression dataset. The IL-17 signaling pathway is upregulated, while MAPK signaling pathway, NOD-like receptor signaling pathway, PD-L1 expression and PD-1 checkpoint pathway in cancer, proteoglycans in cancer, RIG-I-like receptor signaling pathway, and TNF signaling pathway are downregulated. The DEG and GSEA analysis data are presented in Supplemental Tables 3 and 4.
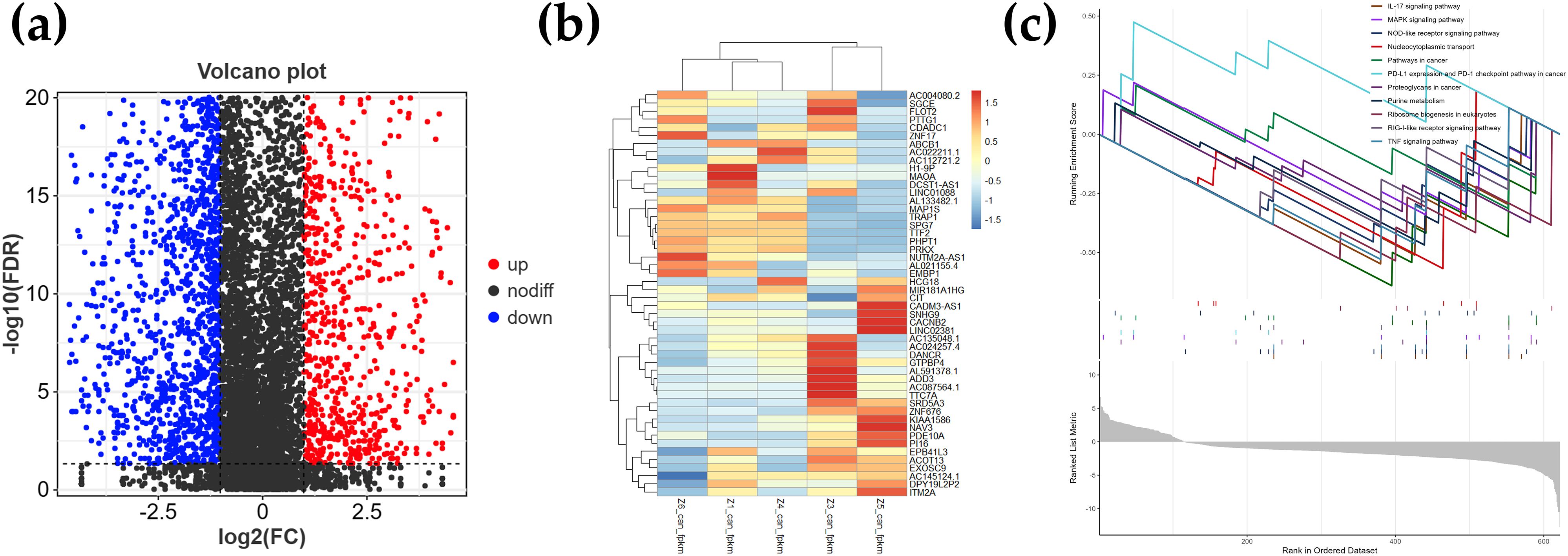
(a) Volcano map of DEGs, which shows the statistically significant expressed genes. The volcano plot shows the fold change against the p value for DEGs. Blue and red dots represent DEGs with significant change (P < .05, FC > 1.5). (b) Heatmap of DEGs. Red and blue colors show the highest and lowest correlation. (c) GSEA analysis of DEGs. GSEA analysis reveals biological mechanism and related pathways. Most pathways such as IL-17 signaling pathway, MAPK signaling pathway, NOD-like receptor signaling pathway are downregulated, and PD-L1 expression and PD-1 checkpoint pathway in cancer is upregulated.
CircRNA Expression Profiling in UCS Patients
Transcriptome sequencing methods was used to systematically profile circRNA expression in all samples. A total of 3403 differentially expressed circRNAs were screened, of which 1650 were up-regulated and 1753 were down-regulated. These results suggest that circRNAs may be involved in the development of UCS through up-regulation or down-regulation of expression, and the volcano map shows that downregulation is slightly dominant. The volcano map and heatmap of circRNA are shown in Figure 3a and b. The circRNAs data is available in Supplemental Table 2.
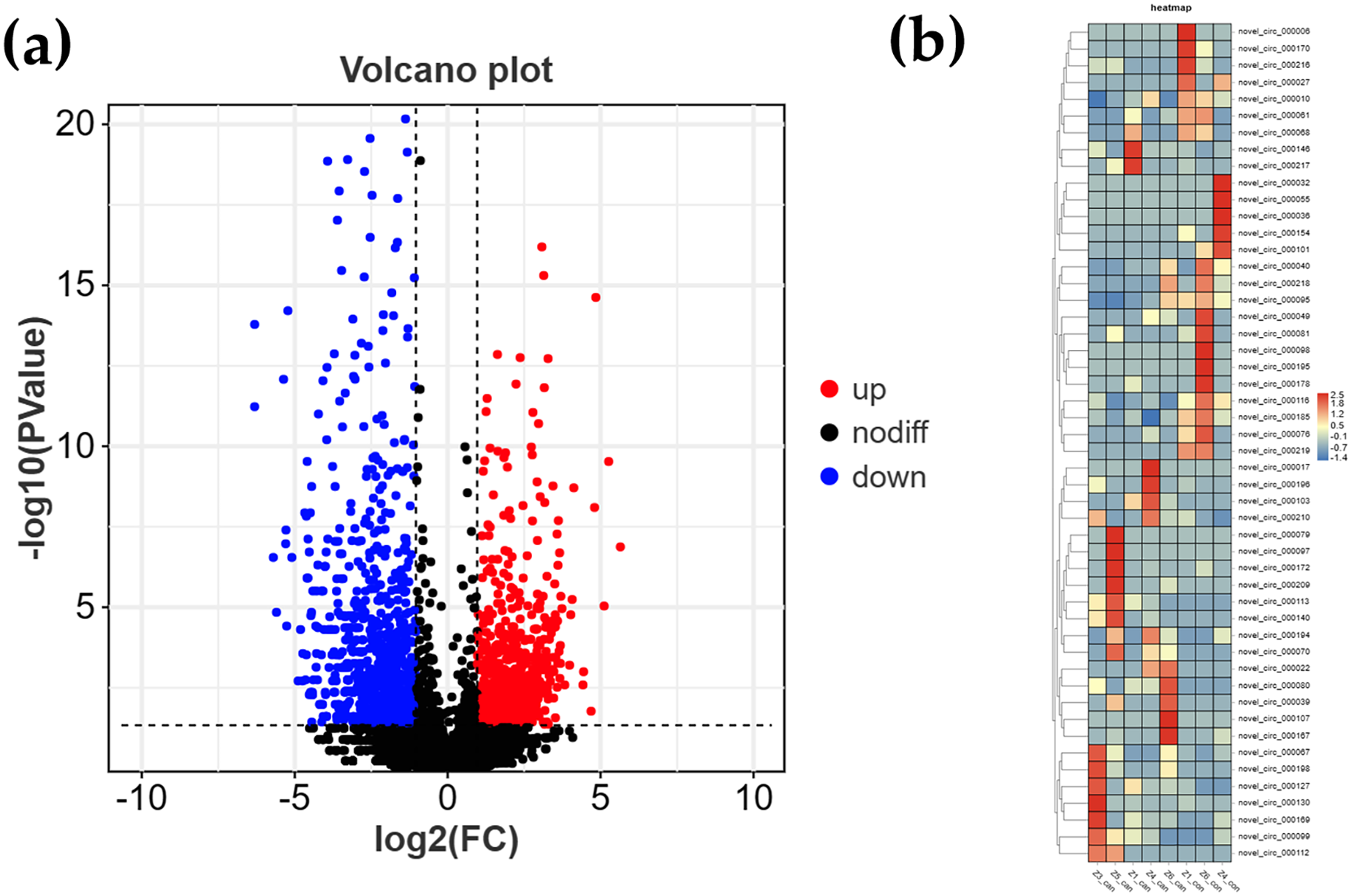
(a) Volcano plot of circRNA. The volcano plot shows the fold change against the p value for circRNAs. Blue and red dots represent circRNAs with significant change (P < .05, FC > 1.5). Volcano plot shows the statistically significant expressed circRNAs. Red dots represent 1650 upregulated circRNAs and blue dots represent the 1753 downregulated circRNAs comparing to paracancerous tissues. (b) Heatmap of circRNA. Red and blue colors show the highest and lowest correlation.
Establishment of miRNA-circRNA-Gene Co-expression Network (CoExpNet)
To further investigate and visualize the correlations of RNAs and genes, a miRNA-circRNA-gene coexpression network was constructed (Figure 4). The pairwise Pearson's correlation coefficient was computed, with 0.99 or above indicating significance. The Pearson correlation coefficient 0.99 indicates a very strong positive correlation between X and Y, that is, Y will increase accordingly as X increases. These significantly correlated miRNAs, circRNAs and genes formed the coexpression network employing the bioinformatics application Cytoscape. The top 5 miRNAs, circRNAs and genes were selected using degree centrality metric in Cytoscape plugin named cytoHubba. These significant miRNAs, circRNAs and genes are highlighted with gradient color according to the their scores. CoExpNet shows ATP9A, HSPG2, DENND5B, UBAP2L and PCNX are the top five related genes in developing UCS, which may become potential targets in UCS treatment. CoExpNet indicates hsa_circ_0002513, hsa_circ_0007797, hsa_circ_0023888, hsa_circ_0023914 and hsa_circ_0034647 are the top five circRNAs, and hsa-miR-485-5p, hsa-miR-6884-5p, hsa-miR-6893-3p, hsa-miR-370-3p and hsa-miR-760 are the top five correlated miRNAs in developing UCS. We reveals corrleations among these genes, miRNAs and circRNAs, and they are significantly related to the pathways and biological behaviors of UCS. Their clinical significance still needs further exploration and verification.
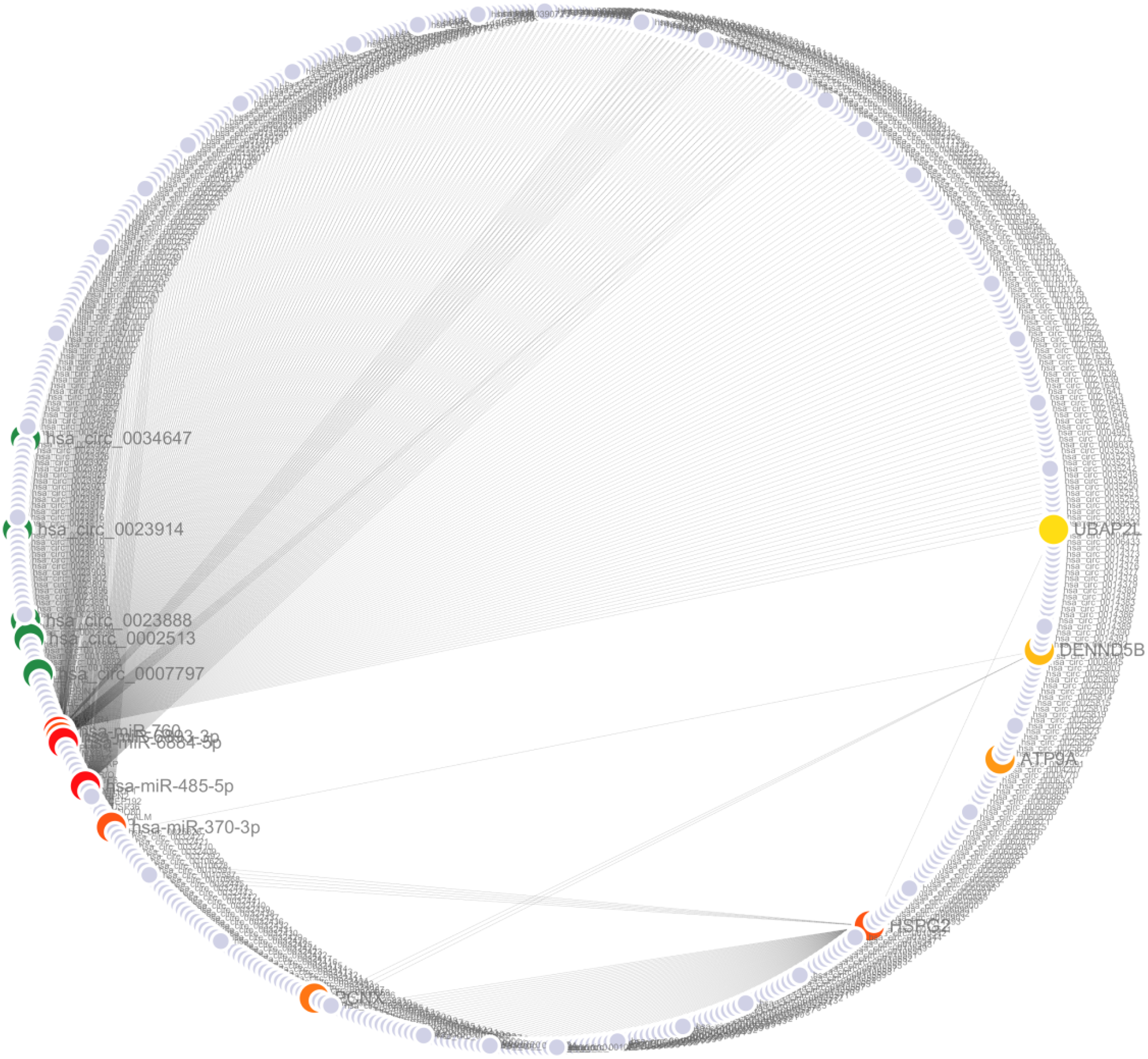
miRNA-circRNA-gene co-expression network (CoExpNet). Co-expression network shows the correlation among miRNA, circRNA and genes. The green thick points represent the top five correlated circRNAs, and the red gradient points represent the top five correlated miRNAs and genes.
Discussion
Our study demonstrates the whole transcriptome results of five UCS patients, including differential expression and distribution of miRNAs, circRNAs, and lncRNAs. We have also developed a novel miRNA-circRNA-gene co-expression network to visualize the interaction of ncRNAs.
Uterine carcinosarcoma (UCS) is a rare, aggressive and lethal gynecological cancer characterized by poor prognosis. Although the pathogenesis and treatment of UCS have been partially studied, the specific mechanism and effective treatment strategies of UCS have not been accurately clarified. In recent years, studies on the mechanism and pathway in tumor by ncRNA have emerged. NcRNAs cannot be translated into protein, but many research show ncRNAs participate in a varity of biological processes. NcRNAs, particularly miRNAs, lncRNAs, and circRNAs, are widely appreciated as pervasive regulators of multiple cancer hallmarks such as proliferation, apoptosis, invasion, and so on. 16 Other effects of ncRNA, such as cancer quiescence, which reflects the ability of cancer cells to enter a reversible slow-cycling or mitotically dormant state and represents a powerful self-protecting mechanism, pave the way for new treatment options. 17 On the basis of the studies above, we use transcriptome sequencing to profoundly visualize RNAs and gene expression in developing UCS via heatmaps, volcano maps and so on. Our research demonstrate the ncRNAs and genes that may be involved in the developing UCS and provide unique insights into potential biomarkers and therapeutic and drug development for UCS.
In this study, we indentified 1632 upregulated and 460 downregulated lncRNAs, 1650 upregulated and 1753 downregulated circRNAs. We firstly construct a miRNA-circRNA-gene coexpression network, and it shows ATP9A, HSPG2, DENND5B, UBAP2L and PCNX are the top five correlated genes in developing UCS. Studies show these genes are related to tumor biological behaviors. ATP9A was critical for regulating macropinocytosis in hepatocellular carcinoma cells, 18 and is an immune checkpoint inhibitor outcome in melanoma and non-small cell lung cancer. 19 GSEA analysis shows the differentially expressed genes (DEGs) mainly involve in IL-7 receptor-based signaling, MAPK signaling pathway, PD-L1 expression and PD-L1 checkpoint pathway and so on, and these pathways have correlations with many tumor-related germinations and progressions. A study shows IL-7 receptor-based signaling is related to the progression of cancer. 20 Additionally, MAPK signaling pathway participates in the activation of pancreatic cancer.21,22 Various cancers express high levels of PD-L1, and the expression of PD-L1 is a significant inhibitor in many cancers such as breast cancer 23 and lung cancer, 24 and it is a potential target in cancer diagnosis and immunotherapy.25–28 CoExpNet also indicates hsa_circ_0002513, hsa_circ_0007797, hsa_circ_0023888, hsa_circ_0023914 and hsa_circ_0034647 are the top five correlated circRNAs, and hsa-miR-485-5p, hsa-miR-6884-5p, hsa-miR-6893-3p, hsa-miR-370-3p and hsa-miR-760 are the top five correlated miRNAs in developing UCS.
We firstly conducted the determination of ncRNAs in the UCS samples, particularly circRNAs. Based on this, we have firstly established the miRNA-circRNA-gene co-expression network of developing UCS. Our study characterized the expressional features of the non-coding RNAs in developing UCS and investigates the plausible pathways of differentially expressed genes in UCS. The pathways may contribute to cancer growth. Five novel genes are identified as possible prognostic predictors and potential therapeutic targets of UCS maligancy, and we reveals the miRNAs and circRNAs related to these genes. To control artificially the type and content changes of RNAs molecules at the cellular level is a new direction for changing UCS's state and drug development. But our study also have many limitations. First, the limited number of samples has led the conclusions not to be generalizable and only to serve as a possible breakthrough. In addition, due to the differences in library preparation methods and the limitation of the amount of sequencing data, the types of one type of ncRNAs obtained by whole transcriptome sequencing are usually fewer than those obtained by specific ncRNA sequencing. This resulted in a discrepancy between the whole transcriptome sequencing results in this study and the actual ncRNAs in the samples. At the same time, differences in the number of patients and age may also affect the generalizability of whole transcriptome sequencing results. For example, the underlying diseases that patients themselves have can also lead to inaccuracies in the inference of UCS biomarkers alone. Next, The findings of our study need to be further validated through experiments to clarify the precise roles of the genes in the development of UCS to provide a definitive direction for clinical research. In all, our findings provide novel ideas for clarifying the mechanisms, achieving early diagnosis and identifying therapeutic targets of UCS maligancy.
Conclusions
In conclusion, our study provides transcriptome sequencing and analyzes results of UCS patients, and we constructed a miRNA-circRNA-gene co-expression network for UCS. Our research innovatively sequences circRNAs and systematically reveals RNAs and genes in this network may be potential diagnostic biomarkers and may play an important role in the onset and development of UCS.
Supplemental Material
sj-xls-1-tct-10.1177_15330338241308607 - Supplemental material for Comprehensive Analysis of Non-coding RNAs Expression Profile and miRNA- circRNA-gene Co-expression Network in Developing Uterine Carcinosarcoma
Supplemental material, sj-xls-1-tct-10.1177_15330338241308607 for Comprehensive Analysis of Non-coding RNAs Expression Profile and miRNA- circRNA-gene Co-expression Network in Developing Uterine Carcinosarcoma by Min-Jun Chen, Ling Yang, Yue Huang, Yunjia Wang, Ying Cai, Chi Zhang, Su-Han Jin, Benjamin Frey, Udo S Gaipl, Yuan Liu, Hu Ma and Jian-Guo Zhou in Technology in Cancer Research & Treatment
Footnotes
Author Contributions
Conceptualization and writing—original draft preparation, Min-Jun Chen, Jian-Guo Zhou; data curation, Ling Yang, Yue Huang, Yunjia Wang; formal analysis, Ying Cai, Chi Zhang; Writing—review and editing, Su-Han Jin, Benjamin Frey, Udo S Gaipl, Yuan Liu, Hu Ma, Jian-Guo Zhou. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
Sample sequencing processes and sequencing results are performed by Omicshare Cloud Platform (Guangzhou, China, https://www.genedenovo.com/). Analysis tools are listed in Methods. Additional data from this study is available in Supplemental Tables 1, 2, 3 and 4. The data for Suining cohort presented in this article are not readily available because the data are part of an ongoing study, due to necessary secrecy. Requests to access the datasets should be directed to the corresponding author.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Statement
This research is approved by Ethics Committee of Zunyi Medical University Affiliated Hospital (Ethics approval number is Zun Yi Lun Shen (2024) 1-028.), and approved by the Review Board of Suining Central Hospital (No. LLSLH20220051).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the National Natural Science Foundation of China, Grant No. 82060475; National Science and Technology Major Project of National Health Commission of China (2023ZD0502105); Chunhui program of the MOE (Ministry of Education of China) (Grant No. HZKY20220231), MOE Liberal arts and Social Sciences Foundation (Grant No. 24YJCZH462), the Natural Science Foundation of Guizhou Province (Grant No. ZK2022-YB632), Youth Talent Project of Guizhou Provincial Department of Education (Grant No. QJJ2022-224), Future Science and Technology Elite Talent Cultivation Project of Zunyi Medical University (ZYSE 2023-02), the Key Program of the Education Sciences Planning of Guizhou Province (Grant No.7), and Collaborative Innovation Center of Chinese Ministry of Education (Grant No. 2020-39). 2021 Zunyi Medical University Graduate Project: ZYK142.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from each participant.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.