
Abstract
Cervical cancer is one of the most malignant tumors in women, particularly those in rural and remote areas. Its underlying molecular mechanisms, including the functions of non-coding RNA (ncRNAs), require more extensive investigation. In this study, high throughput transcriptome sequencing (RNA-seq) was used to identify differentially expressed lncRNAs and mRNAs in normal, cervical intraepithelial neoplasia and cervical cancer tissues from Uyghur women in western China. Dysregulated lncRNAs were found to extensively participate in cervical cancer development, including viral carcinogenesis, cell cycle and cytokine-cytokine receptor signaling. Two miRNA-host lncRNAs, LINC00925 and MIR155HG, showed elevated expression in cervical cancer samples, but prolonged the survival time of cervical cancer patients. The 2 mature miRNAs of the above 2 lncRNAs, miR-9 and miR-155, also showed similar features in cervical cancer. In addition, we identified 545 lncRNAs with potential functions in regulating these 2 miRNAs as competing endogenous RNAs (ceRNAs). In summary, our study demonstrated the dysregulated lncRNAs/miRNAs, particularly LINC00925/miR-9 and MIR155HG/miR-155, regulate the development of cervical cancer by forming a interaction network with mRNAs, highlighting the importance of elucidating the underlying mechanisms of ncRNAs in cervical cancer development.
Keywords
Introduction
Cervical cancer causes an estimated 570,000 cases and 311,000 deaths worldwide, and is the fourth most frequently diagnosed cancer and the fourth leading cause of cancer death in women, 1 particularly in developing countries and rural areas. 2 Screening has indicated that the incidence and mortality rates of cervical cancer are 459–590/100,000 and 17.78/100,000, respectively, for Uyghur women in Xinjiang, China. 3 The limited treatment options for patients with advanced stages of cervical cancer result in a high recurrence rate and poor prognosis. 4 Persistent infection with human papillomavirus (HPV) is the key factor causing cervical cancer. 5 However, most people infected with HPV do not develop cervical cancer, thus, indicating that molecular regulators in host cells are critical in the process of cervical malignant transformation. 6 Numerous studies have demonstrated that cervical cancer is driven by the activation of oncogenes and the inactivation of tumor suppressor genes, which are induced by carcinogens through different mechanisms, including genomic variation, epigenetic regulation and post-transcriptional regulation. 7 -9 In addition, the infection rate of HPV in Xinjiang Uyghur women is lower than that in Han Chinese women. 10 Therefore, the pathogenesis of cervical cancer, including the emerging roles of non-coding RNAs (ncRNAs) in the regulation of gene expression, must be further studied. 11
NcRNAs, previously considered “junk” products of transcription, in fact have important regulatory roles in genome organization, gene expression and various human diseases. 12,13 According to the RNA length, ncRNAs can be artificially classified into long and short groups. 13 Long non-coding RNAs (lncRNAs) are transcripts more than 200 nucleotides in length, which have diverse regulatory mechanisms in cells. 14,15 Accumulating evidence indicates that lncRNAs are involved in malignant processes in cancer, such as tumorigenesis and metastasis, through regulating gene expression. 16 -19 MicroRNAs (miRNAs) are a class of small, endogenous, single-stranded, ncRNA molecules that regulate cell proliferation, differentiation and apoptosis through targeting specific genes. 20 They have also been found to be involved in carcinogenesis and tumor suppression. 21,22 Several dysregulated lncRNAs have been discovered and found to play important roles in cervical cancer. 23 For example, the lncRNA FAM83H-AS1 is regulated by human papillomavirus 16 E6 independently of p53 in cervical cancer cells. 24 The lncRNA DANCR promotes cervical cancer progression by upregulating ROCK1 via sponging miR-335-5p. 25 These findings suggest that more effort is needed to further understand the functions of ncRNAs in cervical cancer.
In recent years, numerous studies have demonstrated an important regulatory competing endogenous RNA (ceRNA) network among different types of RNAs in the cancer development and therapy fields. 26 -28 CeRNA regulatory mechanisms have been implicated in the development of different types of tumors, such as liver, gastric, breast, colon, pancreatic and bladder cancer. 11 A ceRNA network has been constructed to predict a prognostic signature in cervical cancer, comprising 13 messenger RNAs (mRNAs), 2 miRNAs and 3 lncRNAs. 29 In the regulatory network, lncRNAs, pseudogenes, circular RNAs and mRNAs act as ceRNAs, which compete for binding to miRNAs and subsequently regulate their activity. 30 -32 Because of their critical regulatory functions in a wide range of biological processes, lncRNAs have attracted substantial attention among the ceRNAs. 15,33 In cervical cancer, several lncRNAs have been identified as ceRNAs that regulate tumor development, including MIR205HG, 34 the novel lncRNA XLOC_006390 35 and DLX6-AS1. 36 In the ceRNA network, lncRNAs act as molecular sponges that attract miRNAs, thereby contributing to tumorigenesis, cell proliferation, cell apoptosis and metastasis. 37 However, the genomic landscape of cervical squamous cell cancer in Uyghur women in Xinjiang remains to be elucidated.
To further emphasize the important functions of ncRNAs and the ceRNA network in cervical cancer in Uyghur women, we performed transcriptome sequencing to explore the interaction network of lncRNAs, miRNAs and mRNAs in Uyghur women, a minority group in western China. The samples included 3 cervical squamous cell cancer (CSCC), severe cervical squamous intraepithelial lesion (CIN3) and matched normal control (NC) samples. The differentially expressed mRNAs and lncRNAs were obtained to explore their functions in CSCC development and to construct their interaction network through co-expression analysis. We also integrated the roles of miRNAs in the lncRNA-miRNA-mRNA network to identify the ceRNAs in CSCC in Uyghur women, thus, revealing potential regulatory mechanisms of ncRNAs that might serve as drug targets for CSCC in future gene therapy.
Results
Transcriptomic Profile Alterations Were Pronounced in Cervical Squamous Cell Carcinoma Specimens
To explore the transcriptional alterations in cervical cancer, we obtained cervical squamous cell carcinoma stage IIa (CSCC), cervical intraepithelial neoplasia stage III (CIN3, also termed stage 0 carcinoma) and cervicitis tissue (normal control, NC) samples, extracted the RNAs and then performed high-throughput sequencing. Women with CIN3 are at high risk of CSCC. 38 After filtering the low quality reads, we aligned the filtered reads to the human genome sequences with TopHat2. 39 Sample correlation and principal component analysis (PCA) revealed that the 3 CSCC samples were clustered and were distinctly separate from the other 2 groups (Figure 1A, green frame). PCA revealed that the CIN3 samples were closer to the NC samples in the first principal component (Figure 1B). Differentially expressed gene (DEG) analysis (log2|fold change| > 1 and false discovery rate (FDR) < 0.05) was performed to determine the transcriptional differences among the CSCC, CIN3 and NC samples. A total of 1765 and 1372 DEGs were detected from CSCC vs. NC and CSCC vs. CIN3, respectively, whereas only 303 DEGs were detected from CIN3 vs. NC (Figure 1C). By plotting the hierarchical clustering heat map of all DEGs, we found that most of these DEGs showed consistently higher or lower expression in CSCC samples, and more genes were repressed in CSCC samples than in the other 2 groups of samples (Figure 1D). For the upregulated genes in CSCC samples, we observed a clearly increased tendency in CSCC samples, thus suggesting that these genes may regulate CSCC development.

RNA-seq and DEG results. A, Heat map showing the gene expression correlation of all sequenced samples. B, PCA results showing the top 2 principal components explaining the variation in sample distribution. C, Bar plot showing the numbers of up- and downregulated DEGs in the 3 groups. D, Heat map showing the hierarchical clustering results of the DEGs detected in this study. E, Venn diagram showing the co-up- and co-downregulated DEGs in CSCC compared with the other 2 groups. F, Bar plot showing the top 10 enriched GO BP terms for co-upregulated genes in the CSCC group. G, Bar plot showing the top 10 enriched GO BP terms for co-downregulated genes in the CSCC group.
We then analyzed the co-regulated genes in the CSCC group compared with the NC and CIN3 groups. A Venn diagram showed that 226 and 563 genes were co-up- and co-downregulated in the CSCC group, respectively (Figure 1E). We used GO and KEGG pathway enrichment analysis to assess the biological significance of these co-DEGs. We identified the top 10 enriched GO biological process terms for co-upregulated genes in CSCC. The top 5 enriched terms with significant p-values were immune response, inflammatory response, cell-cell signaling, chemotaxis, cell surface receptor signaling pathway, cell proliferation and apoptosis (Figure 1F), which were tightly associated with CSCC in Uyghur patients and may regulate the development of CSCC by altering the tumor microenvironment. For co-downregulated DEGs, mostly homophilic cell adhesion was enriched. Development related terms in other tissues were also enriched (Figure 1G), thus, suggesting that cancer cells may promote tumor development by repressing the expression of these genes.
LncRNAs Are Differentially Expressed Among the 3 Groups
Recent studies have revealed the important regulatory functions of lncRNAs. 15,40 To systematically analyze the expression patterns of lncRNAs in the sequenced samples, we predicted the novel lncRNAs and analyzed their expression patterns. Finally, we predicted 3296 novel lncRNA genes after filtering coding and short transcripts (Table S2). Similar to the results of other studies, the lncRNAs were shorter and had lower expression than the mRNAs (Figure S1A-B). The percentage of exons with length > 200 nt was higher for lncRNAs than mRNAs (Figure S1C). The lncRNAs had fewer exons than the mRNAs (Figure S1D). Sample correlation and PCA results showed that the lncRNA expression profiles in CSCC were more distinct from those of the other 2 groups, as compared with the mRNAs (Figure 2A-B). To explore the differentially expressed lncRNAs (DElncRs), we used FDR < 0.05 and |log2fold change| > 1 as the differential expression cutoff. Compared with NC samples, the cervical cancer tissues contained 746 DElncRs, including 355 upregulated and 391 downregulated lncRNAs. Compared with the CIN3 samples, the CSCC samples contained 662 DElncRs, including 272 upregulated and 390 downregulated lncRNAs. In addition, 197 lncRNAs (118 upregulated and 79 downregulated) were identified as differentially expressed in the CIN3 group compared with the normal group (Figure 2C), results similar to those of the DEGs. Heat maps were used to assess the variations in lncRNA expression (Figure 2C). We found that 153 and 223 lncRNAs overlapped and had the same direction of increased and decreased expression changes, respectively, in the CSCC samples compared with the other 2 groups of samples (Figure 2D). Among the upregulated DElncRs, we selected 4 cervical cancer associated lncRNAs that showed significantly increased expression in CSCC samples: TOPORS-AS1, TMPO-AS1, LINC00925 and MIR155HG (Figure 2E). To verify their expression levels, we obtained 20 additional specimens for each group and performed RT-qPCR for these 4 lncRNAs. All 4 lncRNAs were significantly upregulated in the CSCC samples (Figure 2F,2G), thus suggesting their potential regulatory functions in cervical cancer development. MIR155HG is the host gene of the microRNA miR-155, which is an oncogene. 41,42 LINC00925 is the host gene of miR-9-3p and is considered a cervical cancer-specific lncRNA with significant diagnostic value. 43
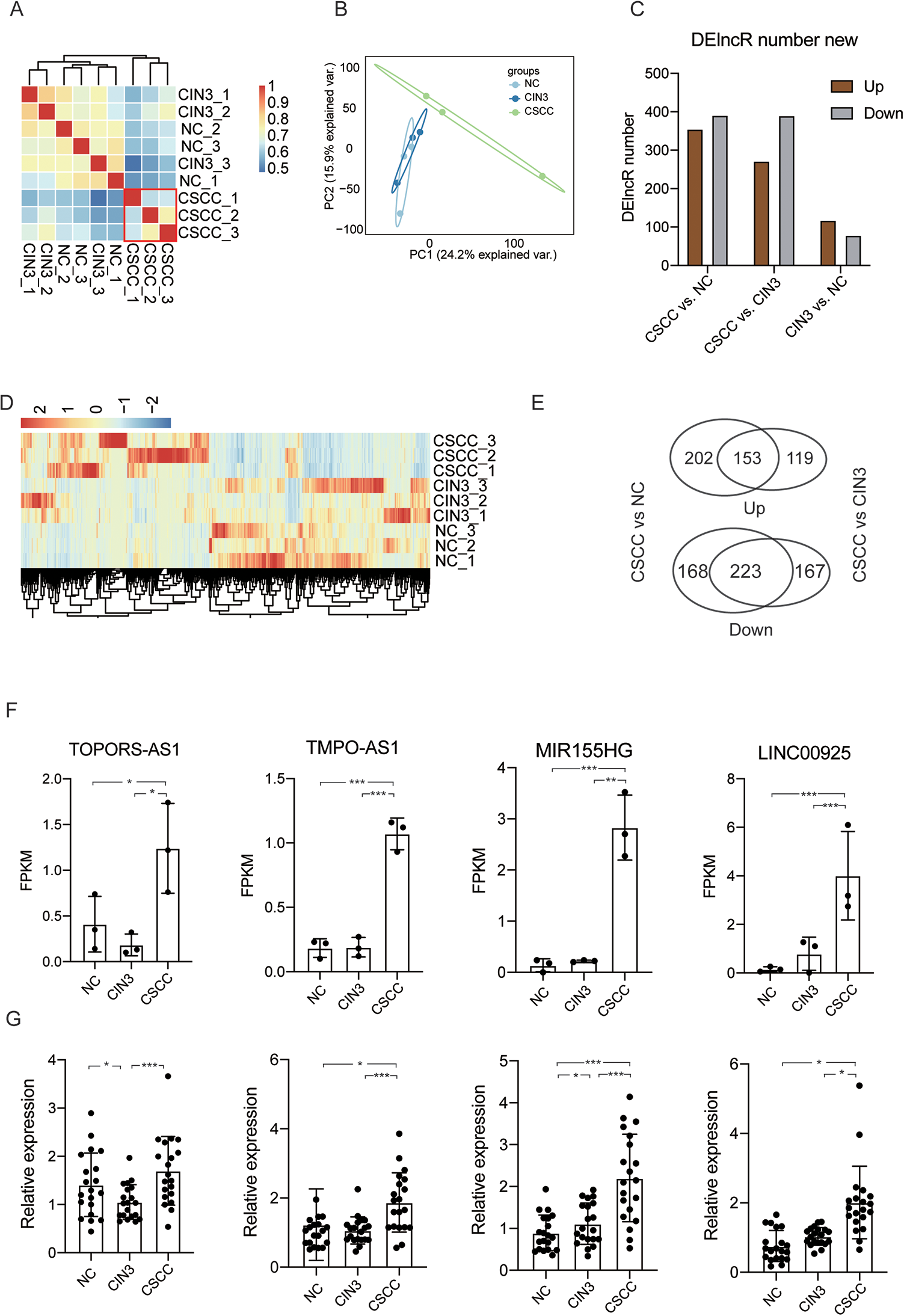
LncRNA expression profiles and DElncR results. A, Heat map of the gene expression correlation showing the distinct expression patterns of lncRNAs in CSCC samples. B, PCA results showing the top 2 principal components explaining the variation in sample distribution. C, Bar plot showing the numbers of up- and downregulated DElncRs in the 3 groups. D, Heat map showing the hierarchical clustering results of the DEGs detected in this study. E, Venn diagram showing the co-up- and co-downregulated DEGs in CSCC compared with the other 2 groups. F, Bar plot showing the expression of 4 selected lncRNAs in the RNA-seq data. G, Bar plot showing the expression of 4 selected lncRNAs in the RT-qPCR experiments.
WGCNA Analysis Revealed Cervical Cancer Associated Differentially Expressed LncRNA Modules
Studies have demonstrated that lncRNAs exert biological functions by regulating neighboring coding genes in-cis or regulating distant genes in-trans. 40 LncRNAs and their target mRNAs often show correlated expression patterns. To further investigate the expression patterns of DElnRs and to construct a lncRNA-mRNA co-expression network, we used the weighted gene co-expression network analysis (WGCNA) method to identify consistent expression modules of DElncRs and DEGs. The expression levels of 1091 DElncRs and 2418 DEGs were combined as input in the WGCNA program. We obtained 11 consistent modules, each represented by a characteristic expression pattern (Figure 3A). Dendrogram clustering of the eigengene values from each module revealed the distinct expression of the 10 detected modules (Figure 3B). We found 4 modules (brown, yellow, green and pink) highly expressed in CSCC samples (Figure 3C-F), which were denoted CSCC specific modules. All CSCC samples in the pink module had high expression, whereas the other 3 modules exhibited higher expression in one sample from the CSCC group. For the above validated lncRNAs, LINC00925 was in the yellow module, MIR155HG was in the green module, and NR2F2-AS1 was in the blue module. The other 6 modules showed an inconsistently higher expression pattern in CIN3 or cervicitis samples. We focused our attention on these 4 cervical cancer related modules.

WGCNA results showing the CSCC specific expression modules. A, Hierarchical clustering dendrogram of all differentially expressed lncRNA and mRNA combined modules. Modules corresponding to branches are indicated according to the colored bands underneath the tree. B, Hierarchical cluster dendrogram and heat map showing the distances of the eigengene values for the modules. C-F, Bar plot showing the distribution of the eigengene values for the 4 CSCC specific modules.
Functional enrichment analysis of mRNAs co-expressed with lncRNAs enables direct exploration of the undetermined functions of lncRNAs. 44 We conducted functional enrichment analysis of co-expressed mRNAs and lncRNAs in these modules and found that viral carcinogenesis functional pathways were in the top 10 KEGG pathways in the pink and yellow modules (Figure 4A and C), including HIST1H4C, HIST1H2BF, CCNE1, PIK3R2, HIST1H4E, CCR8, HIST1H2BM, HIST1H2BO, HIST1H2BL and HIST1H4F. Because the patients with CSCC in this study were infected with HPV, the genes in viral carcinogenesis pathways may play important roles in cervical cancer tumorigenesis and development. Cancer pathways and transcriptional misregulation were detected in the pink and yellow modules, respectively. We also identified the top 10 enriched KEGG pathways in the green module, including cytokine-cytokine receptor interaction, primary immunodeficiency, nitrogen metabolism and linoleic acid presentation (Figure 4B). For the brown module, the cytokine-cytokine receptor interaction pathway was the top pathway (Figure 4D), and the same results were found for the green module (Figure 4B). In the green and brown modules, we also found several immune related modules, including those involved in chemokine signaling pathways, toll-like receptor signaling pathways and antigen processing and presentation. From functional enrichment analysis, we observed that genes with higher expression in CSCC specific modules were associated with tumorigenesis functions (Figure 4E).

WGCNA module functions. A-D, Bar plot showing the top 10 enriched KEGG pathways in CSCC specific modules. E, LncRNA-mRNA network showing the interaction between lncRNAs and mRNAs in the 4 CSCC specific modules. The central purple points represent lncRNAs, and the surrounding points represent mRNAs.
Higher Expression of Host lncRNAs and Mature MiRNAs Was Associated With Prolonged Survival Time in CSCC
On the basis of the 4 validated lncRNAs, we found that 2 lncRNAs (MIR155HG and LINC00925) were the host RNAs of miRNAs (Figure 2F-G). External data analysis from the TCGA database revealed that, although their expression levels were higher in patients with CESC (Figure S2), these 2 lncRNAs significantly prolonged survival time (Figure 5A-B), thus, indicating their anti-cancer function in patients with CC. The mature miRNAs, miR-155 and miR-9-3p, are biomarkers of cervical cancer that show higher expression in cervical cancer tissues. 41 However, survival analysis (with Kaplan-Meier Plotter, http://kmplot.com/analysis/) of the 2 miRNAs revealed their association with prolonged survival times in cervical cancer (Figure 5C-D). Their anti-cancer function contrasted with the cancer promoting functions of miR-155 and miR-9 determined by in vitro experiments. 42,45 Thus, we selected these 2 miRNAs to investigate their functions in CSCC in Uyghur women. We analyzed the functions of the lncRNA co-expressed genes and found that multiple mitotic cell cycle related terms were significantly enriched (Figure 5E-F). To decipher the potential regulatory relationships between host lncRNAs and miRNAs, we overlapped the lncRNA co-expressed genes and miRNA target genes. For MIR155HG and miR-155, only 9 MIR155HG co-expressed genes were potential binding targets of miR-155. For LINC00925 and miR-9, only 20 LINC00925 co-expressed genes were potential binding targets of miR-9, including 11 non-coding RNAs (Figure 5G). Because we selected polyadenylated RNAs to perform RNA-seq, the detected LINC00925 and MIR155HG were not processed into mature miRNAs. These results indicated that MIR155HG and LINC00925 function independently of their processed mature miRNAs.

Functions of 2 selected lncRNAs and their processed mature miRNAs in CSCC. A, Line plot showing overall survival time by classifying patients with cervical cancer into 2 groups on the basis of the expression level of LINC00925. B, Line plot showing overall survival time by classifying patients with cervical cancer into 2 groups on the basis of the expression level of MIR155HG. C, Line plot showing the overall survival time by classifying patients with cervical cancer into 2 groups on the basis of the expression of miR-9. D, Line plot showing the overall survival time by classifying patients with cervical cancer into 2 groups on the basis of the expression of miR-155. E, Bar plot showing the top 10 enriched GO BP terms for genes co-expressed with LINC00925. F, Bar plot showing the top 10 enriched GO BP terms for genes co-expressed with MIR155HG. G, Venn diagram showing the overlapped genes between lncRNA co-expressed genes and miRNA binding targets.
Construction of the ceRNA Network of mRNAs in SCC
To further understand the functions of the 2 selected miRNAs in SCC in Uyghur women, we constructed ceRNA networks composed of mRNAs, lncRNAs and miRNAs. We used TargetScan 46 to predict the potential interacting targets of these 2 miRNAs. We found that the lncRNAs occupied a large portion of the target sets (Figure 6A). This analysis also revealed that 625 genes were targets co-binding miR-155 and miR-9-3p, including 80 mRNAs and 545 lncRNAs (Figure 6B); thus, indicating the competing functions of these target lncRNAs. Functional analysis of these target mRNAs of mature miRNAs revealed that they were enriched in the functional terms, transcriptional regulation, homophilic cell adhesion and negative regulation of cell proliferation pathways (Figure 6C-D). We constructed a ceRNA regulatory network for these 2 miRNAs and their interacting targets, including lncRNAs and mRNAs (Figure 6E). We analyzed the expression patterns of mRNAs in the ceRNA network and found that a large portion were downregulated in CSCC samples (Figure 6F), thus, suggesting that miR-9 and miR-155 repress the expression of target mRNAs. We propose a working model in which the 2 lncRNAs may attenuate cervical cancer development by regulating the expression of cell cycle-related genes. Moreover, miR-9 and miR-155 also attenuate cervical cancer development by repressing cell proliferation and transcription related gene expression (Figure 6G).

CeRNA analysis of 3 oncogenic miRNAs. A, Bar plot showing the potential number of binding targets of 3 miRNAs. The targets were classified into lncRNAs and mRNAs. B, Venn diagram showing overlapping targets between miR-155 and miR-9-3p. The targets of miR-155-5p and miR-155-3p were combined. C, Bar plot showing the top 10 enriched GP BP terms for targets of miR-155-5p. D, Bar plot showing the top 10 enriched GP BP terms for targets of miR-9-3p. E, CeRNA regulatory network showing that lncRNAs function as ceRNAs for 3 oncogenic miRNAs. F, Heat map plot showing the expression patterns of mRNAs involved in the ceRNA network. G, Brief working model showing the lncRNA and miRNA functions in CSCC.
Discussion
Understanding the molecular mechanisms of cervical cancer aids in diagnosis and therapy. 47 The application of ncRNAs in targeted cancer therapy suggests their important regulatory roles in cancers. 48 LncRNAs interact with proteins, miRNAs and mRNAs, forming complex mechanistic networks during tumor pathogenesis and development. 49 In this study, we comprehensively analyzed the transcriptomic changes in CSCC patient samples compared with CIN3 and normal samples. Specifically focusing on lncRNAs, we found that their global expression profile in CSCC was distinct from those in the other 2 groups, thus, suggesting potential biomarker roles in CSCC diagnosis. LncRNA-mRNA co-expression analysis revealed that lncRNAs regulate the cell cycle, viral carcinogenesis and cytokine-cytokine receptor interaction pathways. Finally, we found that 2 lncRNAs had higher expression level in Uyghur CSCC samples but prolonged the survival time in CSCC, thus indicating their rescue function in CSCC. The mature miRNAs processed by these 2 lncRNAs also prolonged the survival time in CSCC. CeRNA analysis indicated the important functional pathways for lncRNAs and miRNAs in CSCC.
Several cervical cancer-associated lncRNAs have been demonstrated to directly bind target proteins or mRNAs and result in post-transcriptional modification. 23 The lncRNA HOXA11-AS is involved in carcinogenesis through regulating the expression of HOxA11. 50 The lncRNA-EBIC is an EZH2-binding lncRNA that represses E-cadherin expression in cervical cancer. 51 Moreover, some studies have suggested that lncRNAs, such as HOTAIR, play significant roles in the progression of cervical cancer by sponging the miRNAs combined with HPV proteins. 52 Pvt1b has been found to act at the intersection of the p53 and Myc transcriptional networks and reinforce the anti-proliferative activities of p53. 53 The lncRNA GIHCG functions as an oncogene and serves as a serum diagnostic biomarker for cervical cancer. 54 Hundreds of our detected DElncRs were upregulated in Uyghur CSCC samples. We validated 4 upregulated lncRNAs by RT-qPCR and found that they showed patterns consistent with the RNA-seq data. Previous studies have shown that these 4 lncRNAs are associated with, and show aberrant expression in, cancers. 55 -58 These results demonstrated that lncRNAs may serve as diagnostic biomarkers for CSCC. However, higher expression of TOPORA-AS1, MIR155HG and LINC00925 is associated with longer survival times in patients with CESC in the TCGA database, possibly because of the differences between in vitro and in vivo experiments, thus indicating the undetected functions of these lncRNAs in cervical cancer.
By constructing an lncRNA-mRNA network, we found that lncRNAs regulate viral carcinogenesis and cytokine-cytokine receptor transduction gene expression through indirect actions, such as functioning as ceRNAs. 59 To construct a ceRNA network, we selected 2 miRNAs associated with cervical cancer. MiR-9 and miR-155 are potential biomarkers for cervical cancer and regulate cervical cancer cell proliferation or epithelial-to-mesenchymal transition. 41,42,60 CeRNAs play important roles in multiple cancers. A previous study has shown that 2 lncRNAs (MIR1281A2HG and OPCML-IT1) and one miRNA (miR-184) are significantly associated with OS in patients with thyroid cancer. 61 MEG3 acts as a cancer suppressor via decreasing the expression of miR-21-5p in cervical cancer in vitro. 62 The expression of TCONS_00026907 is significantly elevated in cervical cancer tissues and significantly promotes cell cycle, proliferation, migration and invasion processes, as well as inhibiting apoptosis by sponging miR-143-5p. 63 HOTAIR promotes cervical cancer invasion and migration by inhibiting the expression of miR-206 and then promoting the expression of MKL1. 64 CeRNA analysis revealed that more than 1000 lncRNAs have the potential to sponge miR-9 and miR-155. Most of these lncRNAs have undetermined functions. By hijacking miRNAs, they indirectly regulate the expression of targeted mRNAs, an aspect requiring further validation. Simultaneously, lncRNAs also regulate biological processes through other actions. 40 Because we have not validated the expression changes in miR-9 and miR-155 in CSCC and normal samples, further exploration of their molecular functions and expression patterns will be necessary in future studies to clarify the underlying mechanisms.
We also detected consistently highly expression of miRNA precursors, which are considered lncRNAs. By analyzing the co-expressed genes for 2 selected lncRNAs, we found little intersection between the lncRNA co-expressed genes and miRNA targets. MiR-9 and miR-155 also prolonged the survival time of patients with cervical cancer, thus, suggesting that they may postpone the development of tumors. The anti-cancer functions of MIR155HG and LINC00925 and their processed mature miRNAs with higher expression in cervical cancer may allow them to attenuate oncogene function through unknown regulatory mechanisms and thus attenuate cervical cancer progression (Figure 6G). Several studies have demonstrated the oncogenic functions of miR-9 and miR-155, 41,42,45 or dual roles of miR-9-5p in cervical cancer. 60 However, all these studies were performed in vitro, and the differences between in vitro and in vivo cellular environments are substantial. Further investigation is needed to decipher how these higher expressed lncRNAs and miRNAs in patients with cervical cancer could prolong the survival time. In cervical cancer, lncRNAs are important as potential biomarkers for cervical cancer prognosis, invasion, metastasis, chemo-resistance and radio-resistance. Our results provide a global overview of deregulated lncRNAs and their potential functional mechanisms in cervical cancer, thus, extending the understanding of lncRNA functions in cervical cancer and providing guidance for future investigations in cervical cancer diagnosis, therapy and molecular mechanisms.
Materials and Methods
Participant Recruitment and Sample Collection
This research was approved by the ethics committee of Xinjiang Medical University. The experiments started in 2014 and ended 2019. All experiments were performed in accordance with the relevant guidelines and regulations. All participants signed an informed consent form voluntarily. The 9 Uyghur women were hospitalized at the First Teaching Hospital of Xinjiang Medical University. Each patient sample was divided into 2 parts: one part was placed in liquid nitrogen within 10 minutes after separation and then stored at −80°C, and the other part was used for pathological diagnosis after hematoxylin-eosin staining. Three control patients had normal cervixes (normal control, NC), and their pathological diagnosis showed chronic cervicitis, 3 patients had severe cervical intraepithelial neoplasia (CIN3), and 3 patients had CSCC (IIa stage). All patients with CSCC and CIN3 had HPV infection. There was no statistically significant difference in age among the 3 groups. Patients with CSCC were newly diagnosed and did not receive radiotherapy or chemotherapy before surgery.
Total RNA was obtained from PBNCs with an RNAprep Pure Blood Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s procedure. The RNA concentration and purity were assessed on the basis of the OD A260/A280 (>1.8) and A260/A230 (>1.6) ratios, and the yield and quality were accessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), with RIN number >7.0.
RNA Library Preparation, Sequencing and Data Processing
For the selected specimens, a total of 10 µg RNA from each specimen was used for directional RNA-seq library preparation. We did not mix specimens during the procedure. Polyadenylated RNAs were purified and concentrated with oligo (dT)-conjugated magnetic beads (Invitrogen, Carlsbad, CA, USA) before library preparation. The purified polyadenylated RNAs were then fragmented with iron at 95°C, then end repaired and ligated with a 5’ adaptor. Reverse transcription was performed with an RT primer with a 3’ adaptor sequence and randomized hexamers to generate complementary DNAs, which were then purified, amplified and stored at −80°C until sequencing. The high throughput sequencing libraries were prepared according to the manufacturer’s instructions. An Illumina NextSeq 500 system was used to sequence the libraries, and 151-nt paired-end sequences were obtained (ABLife Inc., Wuhan, China).
Bioinformatics Analysis
Raw sequencing reads were assessed for potential sequencing issues and contaminants with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). We also trimmed adapter sequences, primers, numbers of fuzzy bases (Ns) and reads with quality scores below 30 with the FASTX-toolkit (version 0.0.13, http://hannonlab.cshl.edu/fastx_toolkit/). Reads that were too short (less than 16 nt) after trimming were also discarded. The quality filtered reads were aligned to the human genome (GRCh38) in TopHat 2.0 software 39 with no more than 4 mismatches allowed. Aligned reads with multiple genomic locations were discarded in the subsequent analysis. After alignment, gene expression levels were obtained by calculation of the fragments per kilobase per million mapped reads.
To analyze the DEGs among the 3 groups, we used the edgeR package to identify DEGs with thresholds of log2|fold change| ≥ 1 and FDR ≤ 0.05.
To systematically investigate the lncRNA features in our RNA-seq data, we used a published lncRNA prediction pipeline to identify novel lncRNAs. 44 Briefly, the transcriptomic data of each sample were assembled separately and then merged to generate a final transcript set with Cufflinks and Cuffmerge. 65 The following processes used to identify lncRNAs were as described by Liu et al. 44 Expression and DElncR analyses of lncRNAs were as described for DEGs.
LncRNA-mRNA Network Construction
The significantly differentially expressed genes and lncRNAs from the pairwise comparisons of the 3 groups were combined into one expression file. WGCNA 66 was performed to obtain the consistently expressed lncRNA-mRNA modules. Pearson correlation coefficients between lncRNAs and mRNAs were also calculated according to the expression levels of DElncRs and DEGs. LncRNA-mRNA pairs with an absolute correlation coefficient value higher than 0.9 and a p-value less than 0.05 were screened.
To predict the binding targets of miRNAs, we used TargetScan v7.0 46 to screen the base pairing regions between mRNAs/lncRNAs and miRNAs. The 3 primer untranslated sequences of mRNAs and the whole lncRNA sequences were used as input.
RT-qPCR Validation
A total of 60 patients were hospitalized at the First Teaching Hospital of Xinjiang Medical University. Among them, 20 control patients had normal cervixes, 20 patients were diagnosed with CIN3, and 20 patients had CSCC. The normal cervical tissues were collected from patients who underwent uterine hysterectomy because of myoma. Patients with CSCC were newly diagnosed and did not receive radiotherapy or chemotherapy before surgery. Patients in the study group with malignant tumors, and those who were pregnant or lactating were excluded. The median age of the patients was 49 years, and there was no significant age difference among the groups.The reaction consisted of one cycle at 95°C for 2 min, followed by 40 cycles of 95°C for 5 sec, and 60°C for 30 sec. Primer sequences are presented in Table S1. Relative quantification was achieved by normalization to the amount of GAPDH with the 2-ΔΔCt method. 67
Other Statistical Methods
To explore the enriched functions of selected gene sets, we used the hypergeometric test to calculate the enrichment of Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Other statistical analysis was performed using R software (R Project for Statistical Computing) unless otherwise stated. The significance of differences was evaluated with either Student’s t-test, when only 2 groups were compared, or the hyper-geometric test for a Venn diagram. *P < 0.05; **P < 0.01; ***P < 0.001. Hierarchical clustering was performed by Cluster 3.0 (http://bonsai.hgc.jp/∼mdehoon/software/cluster/software.htm, in the public domain) or the heat map function in R. Interaction network was generated by Cytoscape software. 68 Survival analysis was performed by GEPIA2 (http://gepia2.cancer-pku.cn/#index) and Kaplan-Meier Plotter (http://kmplot.com/analysis/) for lncRNA and miRNA, respectively.
Data Deposition
The transcriptome sequencing data generated in this study has been deposited in NCBI GEO database with accession ID GSE149763.
Supplemental Material
Supplemental Material, sj-pdf-1-tct-10.1177_1533033821989711 - Dysregulated LncRNAs Act as Competitive Endogenous RNAs and Are Associated With Cervical Cancer Development in UYGHUR Women
Supplemental Material, sj-pdf-1-tct-10.1177_1533033821989711 for Dysregulated LncRNAs Act as Competitive Endogenous RNAs and Are Associated With Cervical Cancer Development in UYGHUR Women by Yanxia Chen, Dong Chen, Jing Wang, Yu Zhang, Ji Zhang, Bing Chen, Yaru Chen, Yi Zhang and Cailing Ma in Technology in Cancer Research & Treatment
Footnotes
Authors’ Note
Yanxia C., C.M. and Yi Z. conceived and designed the experiments. Yanxia C., D.C., Yu Z. participated in the sequence alignment and analyzed the data. J.W., Yaru C., and B.C. performed the validation experiments. Yanxia C., D.C., J.Z. and Yu Z. contribute to figure/dreagents/materials/analysis tools. Yanxia C. and D.C. wrote the paper. All authors read and approved the final manuscript. This research was approved by the ethics committee of Xinjiang Medical University. We confirmed that all experiments were performed in accordance with relevant guidelines and regulations. All participants signed the informed consent voluntarily.
Acknowledgments
Supported by general project of natural science foundation of Xinjiang Uyghur Autonomous Region(2020D01C238), projects of state key laboratory of pathogenesis, prevention and treatment of high incidence diseases in central Asia (SKL-HIDCA-2020-16;SKL-HIDCA-2019-5) and Xinjiang Uyghur Autonomous region science and technology branch (Number: 2019E0282). This study was also supported by ABLife Inc., Wuhan (Number: 201307001).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.