
Abstract
Introduction
RAS gene mutations are prevalent in cancer, with KRAS as the most mutated oncogene. 1 These mutations have been associated with several highly lethal cancers, such as pancreatic ductal adenocarcinoma, non-small cell lung cancer (NSCLC), and colorectal cancer.2,3 Despite the frequency of KRAS mutations, directly targeting KRAS has proved challenging due to its high GTP/GDP affinity and the absence of a deep or pharmacologically accessible small-molecule-binding pocket.4–6 Consequently, research has shifted to indirectly targeting KRAS, including down-stream signaling effectors, epigenetic and synthetic lethality approaches. 2 The challenge of drugging RAS and the urgent need for KRAS targeted therapy in KRAS-driven cancer is thus of great importance to the scientific community. 7
Recently, the discovery of a new allosteric site of KRASG12C has given rise to several irreversible covalently binding inhibitors of KRASG12C, marking a significant leap in KRAS drug development. 8 In 2021, sotorasib (AMG510), a KRASG12C inhibitor, was approved for treating a specific subset of NSCLC patients, 9 and in December 2022, adagrasib (MRTX849), an orally administered potent irreversible KRASG12C mutant inhibitor was approved in the USA. 10 Although these developments represent a significant advance, KRAS inhibitors are far from curative, as tumors can develop resistance due to their plasticity and genetic instability.6,11 Therefore, there is still a need for innovative drug therapies in this field.
The oncogenic KRAS substitution mutation, G12C, has been a challenging therapeutic target in various cancers. LY3499446, a KRASG12C inhibitor, has shown potential as an orally available antineoplastic agent. 12 VT204, a benzothiazole-linked quinazoline compound, was derived from LY3499446 by optimization. In the development of VT204, LY3499446 served as the foundational compound from which further optimizations were derived to enhance its pharmacological profile. VT204 was synthesized by introducing methyl groups at the 2 N-alpha positions of the piperazine ring of LY3499446, aimed at shielding the metabolic sites and thus reducing the overall metabolism of the compounds. This structural modification was specifically designed to improve the stability and bioavailability of VT204 in vivo, facilitating a more consistent therapeutic effect.13–15 Additionally, these modifications are believed to enhance the selectivity for the KRASG12C mutation, thereby reducing potential off-target effects. This optimization process not only underscores the innovative approach taken to refine the pharmacokinetic properties of KRAS inhibitors but also highlights the strategic molecular alterations that can significantly influence the efficacy and safety of targeted cancer therapies. This was achieved through the introduction of a pair of trans methyl groups to the piperazine ring. VT204 has emerged as a promising drug candidate for targeting cancers associated with KRASG12C mutant protein.
In this study, we conducted comprehensive in vivo and in vitro investigations using NCI-H358 cells, which harbor the KRASG12C mutation, and A549 cells, which carry the KRASG12S mutation, as control. Our experiments aimed to demonstrate the effectiveness of VT204 against KRASG12C targets while exhibiting limited activity against KRASG12S, emphasizing its specificity and selectivity. Moreover, our in vivo experiments revealed that VT204 achieved comparable tumor suppression effects to the well-established KRASG12C covalent inhibitor, MRTX849, underscoring its potential as a significant developmental breakthrough in targeted cancer therapy.
Materials and Methods
Reagents and Chemicals
All chemicals and reagents, unless otherwise stated, were procured from commercial suppliers and used without further purification. The compound VT204, depicted in Figure 1A, was synthesized by Suzhou Vincentage Pharma Co., Ltd. The control drug for animal testing, MRTX849 (Figure 1B), was sourced from MCE China (CAS No.: 2326521-71-3). Both compounds, MRTX849 and VT204, underwent assessment of their purity by high-performance liquid chromatography (HPLC), ensuring that both compounds exhibited a purity level of equal to or greater than 98%.

(A) Chemical structure of VT204; (B) chemical structure of KRAS G12C inhibitors MRTX849.
Synthesis of Compound VT204
Detection of reaction processes was done by thin layer chromatography. Column chromatography was performed on silica gel (200-300 mesh). Proton nuclear magnetic resonance (NMR) spectra were obtained using a Bruker Avance 400 MHz NMR spectrometer, and chemical shift values (δ) were reported as parts per million with deuterated dimethyl sulfoxide (DMSO-d6) as the solvent and tetramethylsilane as the internal standard. A Thermo Finnigan HPLC-mass spectrometry (MS)/MS, Surveyor HPLC was equipped with a Diamonsil C18, 4.6 × 250 mm, stainless column and Finnigan LCQ Advantage MAX MS with electrospray ionization (ESI). All solvents and liquid reagents were of analytical reagent grade and were dried in advance and distilled before use. Column chromatography purification was carried out using silica gel. Compound VT204 was synthesized following the 7-step procedure outlined in Scheme 1. The raw data for these characterizations, including NMR spectra, HPLC chromatograms, and mass spectra, can be found in Supplemental Material: Raw Data for Characterization (Supplemental Figures 1 to 4).
General Procedure for Preparation of Compound 2
The amino group of para-2-amino-4-bromo-3-fluorobenzoic acid (substrate 1) (4.0 g, 0.017 mol) and N-chlorosuccinimide (2.5 g, 0.018 mol) were dissolved in N, N-dimethylformamide (DMF) (40 ml), heated to 70 °C, and reacted overnight. The reaction was poured directly into 100 ml of ice water and the chlorinated substituent 2-amino-4-bromo-5-chloro-3-fluorobenzoic acid (compound 2) (4.6 g) was obtained by filtration.
General Procedure for Preparation of Compound 3
Compound 2 (4.6 g, 0.017 mol) was dissolved in ethanol (60 ml) and then undergoes a cyclization reaction with formamidine acetate (21.4 g, 0.2 mmol) by heating and refluxing overnight, resulting in ring closure to form 7-bromo-6-chloro-8-fluoroquinazolin-4-ol (compound 3). After the reaction was complete, the dry ethanol was concentrated, 20 ml of water and 60 ml of ethyl acetate (EA) were added, and the liquid was partitioned and dried. The organic phase was concentrated to obtain a crude solid (5.2 g), which was used directly in the next step.
General Procedure for Preparation of Compound 4
Compound 3 (5.2 g, 0.018 mol) was added to SOCl2 (30 ml) with 6 drops of DMF and heated to reflux overnight to give 7-bromo-4,6-dichloro-8-fluoroquinazoline (compound 4). After the reaction was complete, the dry SOCl2 was concentrated, saturated sodium bicarbonate and EA were added, the liquid was partitioned, the organic phase was dried and concentrated, and purified by column chromatography to give a yellow solid (3.0 g).
General Procedure for Preparation of Compound 6
Subsequently, the piperazine analog (2R,5S)-tert-butyl 2,5-dimethylpiperazine-1-carboxylate (compound 5) was added to undergo the Nucleophilic Aromatic Substitution reaction (SNAr). Compound 4 (296 mg, 1.0 mmol) and compound 5 (400 mg, 1.9 mmol) were dissolved in dioxane (6 ml), N, N-diisopropylethylamine (387 mg, 3.0 mmol) was added, and the reaction was heated to 50 °C. At the end of the reaction, H2O (5 ml) and EA (15 ml) were added, the organic phase was extracted, dried and concentrated, and purified by column chromatography to give the tert-butyl (2R,5S)-4-(7-bromo-6-chloro-8-fluoroquinazolin-4-yl)-2,5-dimethylpiperazine-1-carboxylate (compound 6) (300 mg).
General Procedure for Preparation of Compound 8
Compound 6 (200 mg, 0.42 mmol) was dissolved in dioxane/H2O (6 ml/2 ml), boric acid (compound 7) (163 mg, 0.52 mmol) and 1,1′-bis (di-t-butylphosphino) ferrocene palladium dichloride (PdCl2(dtbpf)) (28.4 mg, 0.04 mmol, 0.1 eq), K3PO4 (139 mg, 0.65 mmol) for Suzuki coupling reaction, and heated under N2 protection to 90 °C for 2 h. At the end of the reaction, 5 ml of water and 15 ml of EA were added and the organic phase was extracted, dried and concentrated, and purified to give a solid compound 8: tert-butyl (2R,5S)-4-(7-(2-((tert-butoxy carbonyl) amino)-7-fluorobenzo[d]thiazol-4-yl)-6-chloro-8-fluoroquinazolin-4-yl)-2,5-dimethylpiperazine-1-carboxylate (78 mg).
General Procedure for Preparation of Compound 9
Compound 8 (78 mg, 0.12 mmol) was dissolved in di-chloromethane (6 ml), 1 ml of trifluoroacetic acid (TFA) was added, and the reaction was carried out overnight at room temperature. The aim was to deprotect the Boc group of compound 8 under TFA conditions, leading to the formation of 4-(6-chloro-4-((2S,5R)-2,5-dimethylpiperazin-1-yl)-8-fluoroquinazolin-7-yl)-7-fluorobenzo[d]thiazol-2-amine (compound 9); add EA, adjust pH = 8 with saturated sodium bicarbonate, dry and concentrate the organic phase to obtain the crude product (70.0 mg).
General Procedure for Preparation of Compound VT204
Finally, acryloyl chloride selectively acylated the nitrogen atom of piperidine, producing the target compound VT204. Specific reactions were as follows, compound 9 (70.0 mg, 0.152 mmol) was dissolved in tetrahydrofuran/H2O (2 ml/2 ml), K2CO3 (95.3 mg, 0.69 mmol) was added, cooled to 0 °C under N2 protection, acryloyl chloride (14.2 mg, 0.156 mmol) was added slowly dropwise, and the reaction continued after 0 °C for 10 min. The reaction was completed by adding water, extracting with EA, drying the organic phase, concentrating and purifying by column chromatography to obtain a white solid (30.0 mg).
Molecular Docking Study
Molecular modeling studies were conducted using the Schrödinger Molecular Modeling Suite 2011 (Schrödinger LLC, New York, NY, USA) running on a Windows work-station. The KRASG12C protein structure (PDB: 6UT0) was procured from the RCSB Protein Data Bank (http://www.rcsb.org/pdb). VT204 and MRTX849 were prepared using the LigPrep module and docked into the SWII pocket following the covalent docking protocol in the Schrödinger Suite.
Cell and Animal Models
Human lung cell lines NCI-H358 (CL-0400) and A549 (CL-0016), bearing KRAS mutations, were purchased from Procell Life Science & Technology Co., Ltd. The normal human lung epithelial cell line BEAS-2B was obtained from the American Type Culture Collection (Manassas, VA, USA). NCI-H358 and A549 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 (Gibco) and BEAS-2B cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, 8121211), both media were supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% antibiotics (penicillin/streptomycin) (Gibco) at 5% CO2 at 37 °C. The medium is refreshed every 2 to 3 days. Upon reaching 90% confluence, the old medium is aspirated, and trypsin is added for cell detachment and subsequent subculture. BALB/c nude male mice (aged 6-8 weeks) were sourced from the CAVENS Laboratory Animal Co., Ltd, and housed in a standard laboratory condition (temperature 22 ± 2 °C and humidity 50%-60%). Mice arrive at the laboratory with an acclimatization period of 7 days. The study was approved by the Ethics Committee of the Department of Laboratory Animal Science at Guilin Medical University, with the approval number GLMC-IACUC-2023004. This animal experiment followed the blind method. Additionally, all animal experiments were carried out in accordance with the “Guide for the Care and Use of Laboratory Animals, 8th Edition,” 16 and the reporting of this study conforms to the ARRIVE 2.0 guidelines. 17 These measures ensure compliance with ethical standards for the humane treatment of animals in research, emphasizing efforts to minimize animal use and alleviate suffering throughout the study. This animal experiment followed the blind method.
Cell Viability Assay
Cell viability of NCI-H358 cells harboring the KRASG12C mutation and A549 cells with the KRASG12S mutation was evaluated using the Cell Counting Kit-8 (CCK8) assay (Beyotime, Shanghai, China). A total of 3 × 103 cells were seeded in 96-well plates and allowed to adhere for 24 h. Cell viability was assessed with CCK8 solution following 0 , 24, and 48 h incubation with VT204. Absorbance was measured at 450 nm on a microplate reader (Varioskan Flash, Thermo Scientific, USA) after a 2 h incubation at 37 °C, and the IC50 was calculated using GraphPad Prism. All subsequent experiments utilized the IC50 concentration established for NCI-H358 cells. To test toxicity on normal cells, BEAS-2B cells were seeded in 96-well plates and treated and the processing and detection methods were the same as described in NCI-H358 and A549 cells.
Colony Formation Assay
The clonogenic capacity of NCI-H358 and A549 cells, both carrying different KRAS mutations, was evaluated post VT204 treatment. A total of 500 cells were seeded in 6-well plates and allowed to adhere for 24 h before treatment with VT204 at indicated concentrations, alongside a vehicle control. After a 24 h treatment with VT204, the cells were washed with PBS and then cultured in fresh medium without VT204 for a period of 7 days to allow the formation of colonies. Throughout this period, the medium was refreshed every 2 days to maintain optimal cell conditions. This colony formation assay was conducted to assess the long-term impact of VT204 on cell growth and proliferation. Finally, colonies were fixed with 4% paraformaldehyde and stained with 4% crystalline violet (Solarbio) for 10 min. Colony numbers were determined through direct visual inspection.
Cell Cycle Analysis
NCI-H358 and A549 cells were seeded in 6-well plates and treated with VT204 for 24 h. Post-treatment, cells were collected, fixed with 70% ethanol, and then resuspended in propidium iodide (PI) staining solution (Beyotime). For each sample, 10 000 events were analyzed using flow cytometry (MACSQuant, Miltenyi Biotec GmbH), and the proportions of cells in different cell-cycle phases were determined using the ModFit LT software.
Annexin V-FITC Apoptosis Assay
Apoptotic cells induced by VT204 treatment were identified via flow cytometry (MACSQuant, Miltenyi Biotec GmbH). After VT204 treatment, NCI-H358 and A549 cells were detached using EDTA-free trypsin, followed by apoptosis assessment using the Annexin V-FITC Apoptosis Detection kit (Vazyme). A minimum of 10 000 events were collected and analyzed for Annexin V-FITC staining. The proportion of Annexin V-positive apoptotic cells was calculated using FlowJo™ v10.8 Software.
Wound Healing Assay
Confluent (90%-95%) cultures of NCI-H358 and A549 cells in 6-well plates were scratched using sterile pipette tips. The treatment group cells were exposed to VT204 for 24 h before being rinsed with PBS, while control cells were cultured in RPMI 1640 medium supplemented with 1% FBS. Scratch areas were photographed at 0, 24, and 48 h, and imaged under a microscope. Cell migration rates were measured using ImageJ software.
Transwell Assays
NCI-H358 and A549 cells were treated with VT204 for 24 h, then resuspended at a density of 1 × 105 cells/well (200 μl) and seeded in the upper chambers of 24-well migration chambers (8.0 μm pore membrane, Corning, New York, USA) filled with serum-free medium. Chambers were precoated with Matrigel (Corning) for cell invasion studies. The lower chambers were filled with 700 μl medium supplemented with 10% FBS. After a 24 h incubation, cells were fixed, stained, and imaged under a microscope. The number of invaded cells was quantified using ImageJ software.
Protein Extraction and Western Blotting
Total proteins were isolated from cells and tumor tissues using radioimmunoprecipitation assay buffer fortified with 1% protease inhibitors, followed by quantification using a bicinchoninic acid protein assay kit (Thermo Scientific). Proteins were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto polyvinylidene fluoride membranes (Sigma), blocked with bovine serum albumin (Sigma), and incubated with the primary antibody overnight at 4 °C. After incubation with horseradish peroxidase-conjugated secondary antibodies for 1 h, proteins were detected using an enhanced chemiluminescence kit (Thermo Scientific). The photos were taken using the Bio-Rad Gel Imager, and the blots were analyzed using ImageJ software. GAPDH (60004-1-lg, proteintech, 1:20 000) was used as the loading control. The primary antibodies used were rabbit anti-phospho-p44/42 MAPK (Erk1/2) (#4370, Cell Signaling Technology, 1:2000) and rabbit anti-p44/42 MAPK (Erk1/2) (#4695, Cell Signaling Technology, 1:1000). The secondary antibodies used were goat anti-rabbit (A0545, proteintech, 1:10 000) and goat anti-mouse (A9044, proteintech, 1:10 000).
Tumor Growth Inhibition Analysis
The BALB/c nude male mice were randomly divided into 2 groups, which were subcutaneously inoculated on their right flanks with NCI-H358 and A549 cells (5 × 106 cells in 100 μl serum-free RPMI 1640, mixed with 50% Matrigel), respectively. Tumor growth was monitored until they reached a size of approximately 200 to 250 mm3. Mice bearing NCI-H358 cells were then arbitrarily segregated into 3 groups (n ≥ 6 per group): a vehicle control group, a VT204 treatment group, and an MRTX849 treatment group. Similarly, mice harboring A549 cells were divided into 2 groups (n ≥ 6 per group): a vehicle control group and a VT204 treatment group.
Tumor growth inhibition (TGI) was assessed using subcutaneous xenograft models of NCI-H358 and A549 cells in BALB/c nude mice. Mice were randomly divided into treatment and control groups. VT204 and MRTX849 were prepared in a solution containing 0.5% (w/v) methyl cellulose and 0.5% (w/v) Tween 80 in water, which also served as the vehicle control. Treatments were administered orally at a dosage of 50 mg/kg on alternate days. Tumor dimensions and body weights were recorded thrice weekly and daily, respectively. Upon conclusion of the experiment, mice were euthanized, and tumors were harvested for further analysis.
Tumor volume was estimated as (width2 × length/2), where width is the smaller dimension and length is the larger one. TGI was calculated using the formula:

In Vivo Safety Evaluation
To evaluate the safety of VT204 in mice, euthanized mice were subjected to tissue fixation using 4% paraformaldehyde. Specifically, heart, liver, spleen, lung, and kidney tissues were carefully collected. Subsequently, these tissues underwent a dehydration process using graded sucrose solutions and were embedded in paraffin wax. Thin sections of approximately 5 µm thickness were obtained from the embedded tissues. Following this, the sections were subjected to staining with hematoxylin–eosin (H&E) to facilitate histopathological examination. To visualize the tissue samples, an optical microscope (Nikon) was utilized to capture images.
Statistical Analysis
Data were analyzed using GraphPad Prism 9.5.1 and are represented as mean ± standard error of mean (SEM). Comparison between 2 groups was done using an unpaired 2-tailed Student's t-test. For multiple groups, a 1-way analysis of variance (ANOVA) or 2-way repeated measures ANOVA was employed. A P-value < .05 was considered statistically significant.
Results
The Structures of the Compound VT204 was Characterized by 1H NMR, 13C NMR and MS Studies
The compound 1-((2R,5S)-4-(7-(2-amino-7-fluorobenzo[d]thiazol-4-yl)-6-chloro-8-fluoroquinazolin-4-yl)-2,5-dimethylpiperazin-1-yl) prop-2-en-1-one (VT204) was prepared as a white solid. The NMR and ESI MS characterization data are provided as follows: 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.93 (d, J = 17.5 Hz, 3H), 7.27 (ddd, J = 12.9, 8.4, 5.7 Hz, 1H), 7.08 (t, J = 8.8 Hz, 1H), 6.95 to 6.66 (m, 1H), 6.18 (dd, J = 16.6, 2.0 Hz, 1H), 5.74 (d, J = 9.9 Hz, 1H), 4.61 (d, J = 128.2 Hz, 2H), 4.20 to 3.77 (m, 4H), 1.34 to 1.13 (m, 6H). 13C NMR (101 MHz, DMSO) δ 167.46, 167.40, 165.14, 164.89, 162.62, 157.72, 155.28, 154.31, 154.12, 153.46, 140.78, 128.28, 127.68, 120.19, 116.79, 116.63, 107.18, 106.98, 50.69, 48.71, 48.39, 48.07, 15.83, 14.72. MS m/z (ESI): 515.1 [M + 1]. HPLC: tR 7.525 min, purity 99.259%.
Molecular Docking Study
To explore the targeting of VT204 to MRTX849 even further, by analyzing the active pockets of KRASG12C-VT204 and KRASG12C-MRTX849 (Figure 2A and B), it was found that VT204 binds to the same pocket as MRTX849, with a docking score of −9.098, indicating a similar binding affinity to MRTX849's score of −9.496. VT204 forms key polar interactions, including hydrogen bonds with ASP69 and GLU63, halogen bonds with TYR64 and HIS95, Pi-action with ARG68, and Pi–Pi stacking with TYR96 (Figure 2C). In contrast, MRTX849's interactions include salt bridges and H-bonds with ASP92, GLU62, and TYR96, and H-bonding with LYS16 (Figure 2D). These results suggest that VT204 may serve as a potent KRASG12C inhibitor with efficacy comparable to MRTX849.
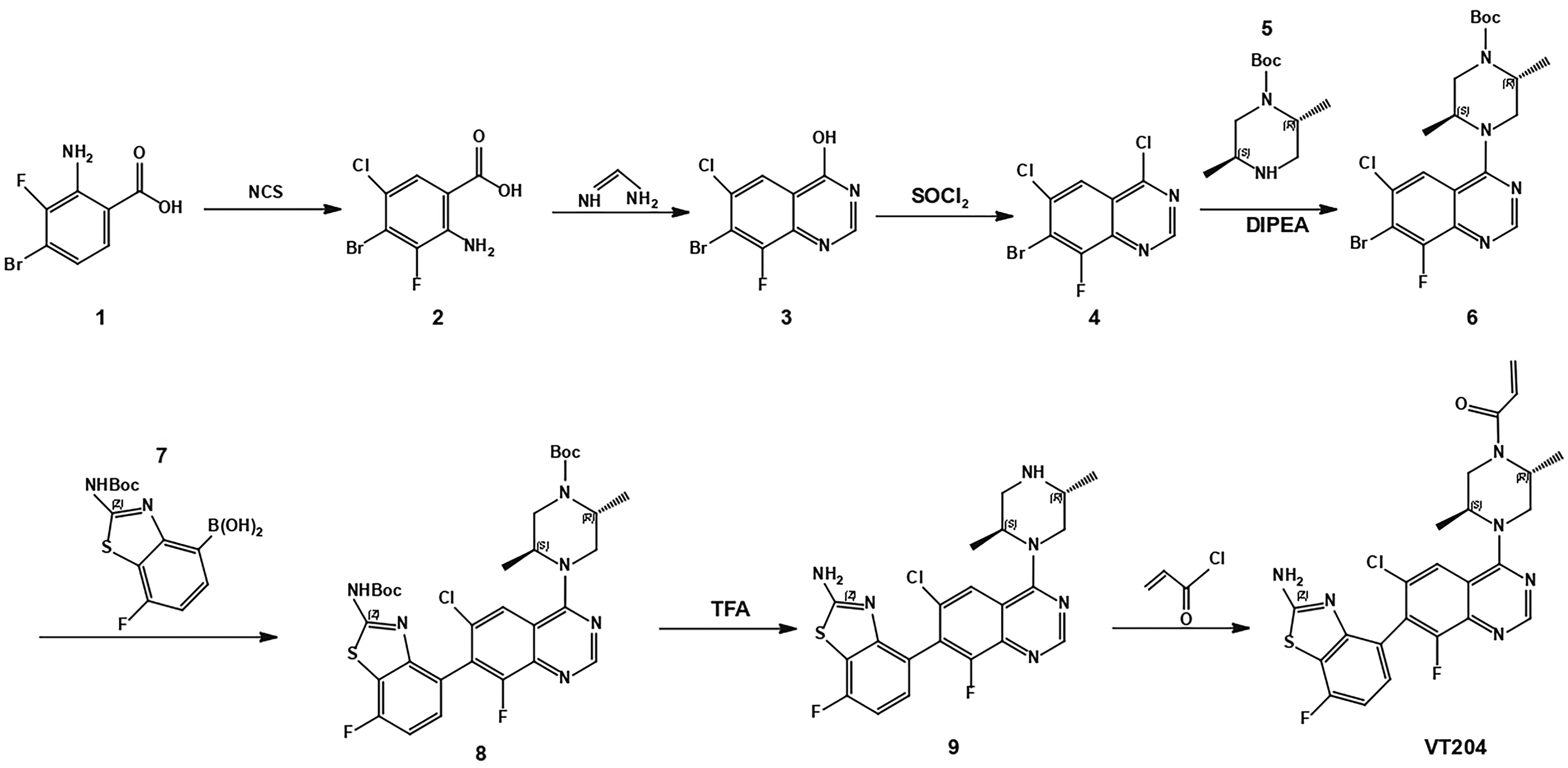
Synthesis of compounds VT204.
VT204 Suppressed Cell Proliferation in NSCLC Cells
The effect of VT204 on cell viability and proliferation in NSCLC was assessed using A549 and NCI-H358 cells. Cells were treated with various concentrations of VT204 for 24 or 48 h. The CCK8 assay revealed that VT204 inhibited cell viability in a concentration- and time-dependent manner (Figure 3A). Treatment with VT204 at concentrations of 8 and 3 μM significantly reduced cell viability in NCI-H358 cells after 24 and 48 h, respectively. In contrast, VT204 did not significantly inhibit A549 cell viability until 24 and 48 h of treatment at concentrations of 38 and 13 μM, respectively. Based on its specific targeting of KRASG12C, a concentration of 8 μM was chosen for further experiments. The colony formation assay demonstrated that VT204 effectively inhibited the growth of NCI-H358 cells at a concentration of 8 μM but had limited impact on A549 cells (Figure 3B). CCK8 was used to determine the effect of VT204 on the viability of BEAS-2B cells in vitro. The results showed that the viability of BEAS-2B cells did not change significantly (Supplemental Figure 5). This showed that at an appropriate dose VT204 normal cells are virtually non-toxic.

(A) Structure model of VT204 in KRASG12C; (B) structure model of MRTX849 in KRASG12C; (C) 2D model of VT204; (D) 2D model of MRTX849.
VT204 Suppressed the Migration and Invasion Capacities of NSCLC Cells
To evaluate the effect of VT204 on the metastatic potential of NSCLC cells, wound healing and transwell assays were conducted. Treatment with VT204 significantly reduced wound healing in NCI-H358 cells compared to the control group (Figure 3C). The scratch closure rates of VT204-treated NCI-H358 cells at 24 and 48 h were 9.99% and 6.34%, respectively, which were significantly lower than those of the control group (27.57% and 75.63%).
In contrast, VT204-treated A549 cells showed no significant difference in scratch closure rates at 24 h (32.98%) and 48 h (63.32%) compared to the control group (39.76% and 65.59%). These results indicate that VT204 specifically inhibited migration in KRASG12C-targeted NCI-H358 cells. The transwell assay further confirmed the inhibitory effect of VT204 on cell invasion, with significant inhibition observed in NCI-H358 cells (invasion rate of 55.04%), while no significant difference was observed in A549 cells (invasion rate of 94.57%) (Figure 3D).
VT204 Induced G2/M-Phase Arrest in NCI-H358 Cells
To investigate the mechanism underlying the inhibition of NSCLC cell proliferation by VT204, cell cycle distribution analysis was performed. In Figure 4A, it can be observed that the percentage of cells in the G2/M phase increased from 6.78% to 16.59% after treatment with 8 μM VT204 for 24 h, suggesting the induction of G2/M-phase arrest in NCI-H358 cells. Simultaneously, there was a decrease in the number of cells in the G0/G1 phase. This finding indicates that VT204 treatment affected the cell cycle distribution of NCI-H358 cells, leading to the accumulation of cells in the G2/M phase and a reduction in the proportion of cells in the G0/G1 phase. In contrast, no significant differences in cell cycle distribution were observed in A549 cells compared to the control group.
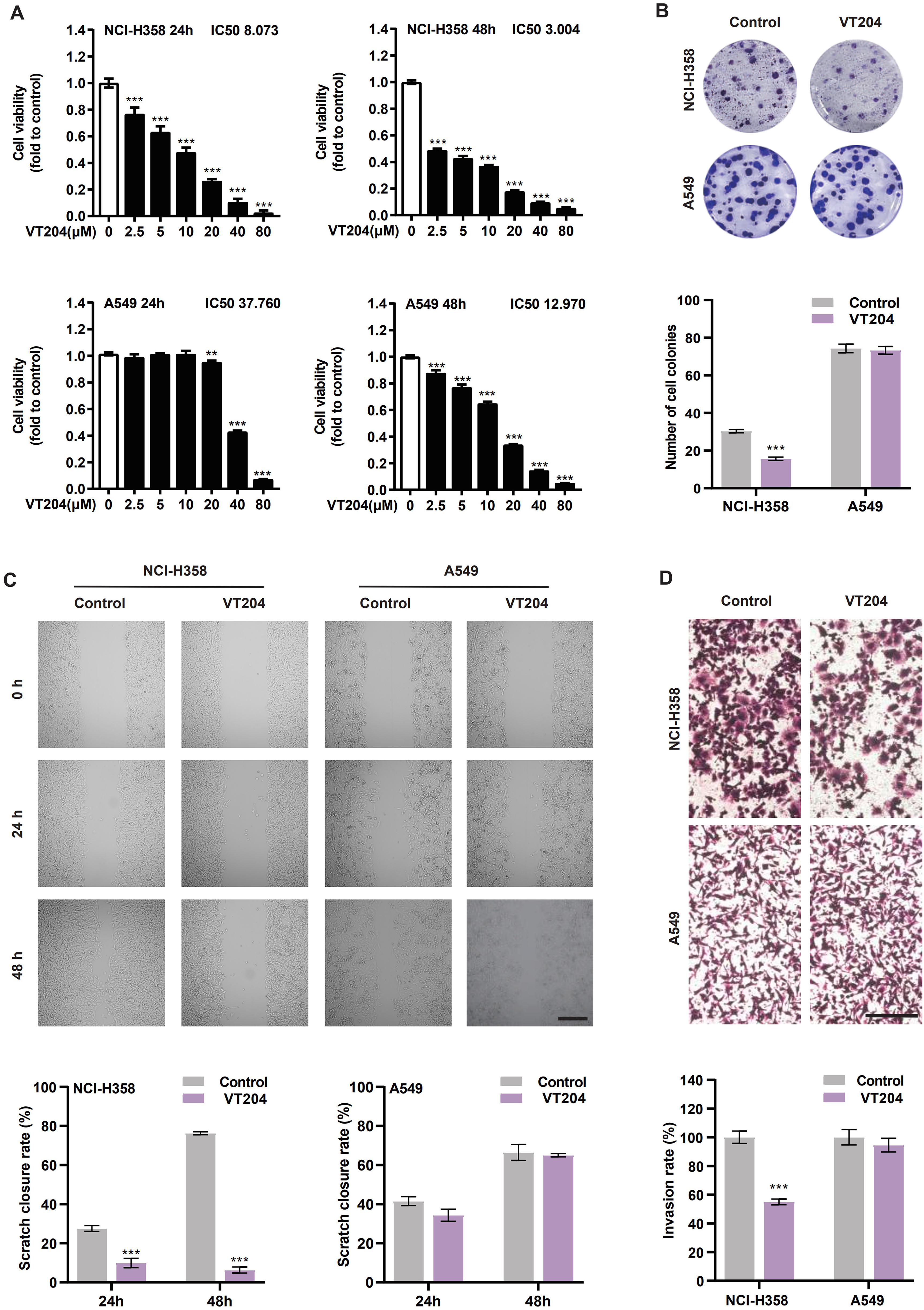
(A) CCK8 assay of cell viability in NCI-H358 and A549 cells after VT204 treatment. (B) Plate colony formation of NCI-H358 and A549 cells after VT204 treatment for 7 days and quantitation of colony numbers. (C) Typical images of NCI-H358 and A549 cells treated with 8 μM VT204 for 24 h in the wound healing assay to evaluate migration capacity. Scar bar = 200 μm. (D) Transwell assays with Matrigel were performed to evaluate the invasion capacity. Scar bar = 100 μm. All the data shown represent results of 3 individual experiments (n = 3). Data were mean±SEM. ***P < .001.
VT204 Induced Apoptosis in NCI-H358 Cells
In consideration of the pivotal role of apoptosis as a major mechanism for the antiproliferative effects of anticancer drugs, we investigated the potential of VT204 to induce apoptosis in both NCI-H358 and A549 cells. Flow cytometry analysis demonstrated that treatment with 8 μM VT204 resulted in a notable increase in the proportion of apoptotic cells in NCI-H358 cells, reaching 23.80%, which was significantly higher than the control group (12.54%) (Figure 4B). However, in contrast to NCI-H358 cells, VT204 did not induce significant apoptosis in A549 cells. These findings suggest that VT204 selectively induces apoptosis in KRASG12C-targeted NCI-H358 cells, providing valuable insights into its mechanism of action in inhibiting NSCLC cell growth through apoptosis induction.
VT204 Functions by Modulating Erk Signaling
Given the well-established knowledge that growth factor-mediated activation of KRAS leads to the activation of the RAF/MEK/ERK pathway, a critical downstream signaling pathway of KRAS, 18 we aimed to investigate the impact of VT204 on the phosphorylation status of extracellular signal-regulated kinase (ERK) in both NCI-H358 and A549 cells. The Western blot analysis was employed to elucidate whether the inhibition of cell viability in NCI-H358 and A549 cells by VT204 is reliant on KRAS signaling.19,20
The results revealed that treatment with VT204 effectively suppressed the phosphorylation of ERK1/2 (pErk) in NCI-H358 cells (Figure 4C), while it had no significant effect on the total levels of ERK1/2. These findings suggest that VT204 inhibits the growth and survival of NCI-H358 cells through the modulation of KRAS downstream phosphosignaling. In contrast, VT204 did not exhibit a similar impact on A549 cells, indicating a selective and specific inhibitory effect of VT204 on KRASG12C-targeted NCI-H358 cells. These observations shed light on the underlying mechanism by which VT204 exerts its antitumor effects in NSCLC cells by regulating KRAS-mediated signaling pathways.
VT204 Inhibited the Growth of NSCLC Cells In Vivo
The in vivo antitumor efficacy of VT204 was assessed using subcutaneous xenograft models of NCI-H358 and A549 cells in BALB/c nude mice (Figure 5A). After subjecting NCI-H358 xenograft-bearing mice to 23 days of treatment with VT204, a remarkable reduction in tumor volume was observed compared to the control group (Figure 5B and C). The TGI achieved by VT204 was calculated to be 77.43%, which is notably similar to the TGI achieved by MRTX849 (85.14%). These findings indicate that VT204 possesses significant antitumor efficacy in vivo, comparable to the well-established KRASG12C inhibitor MRTX849. The substantial tumor volume reduction (P<.001 vs control) observed in the VT204-treated group highlights the promising potential of VT204 as a therapeutic agent for targeting KRASG12C-driven tumors. The close resemblance of TGI between VT204 and MRTX849 further supports the notion that VT204 exhibits comparable inhibitory effects on KRASG12C-induced tumor growth. These results reinforce the relevance and significance of VT204 as a promising candidate for further development as a KRASG12C inhibitor in the treatment of NSCLC.
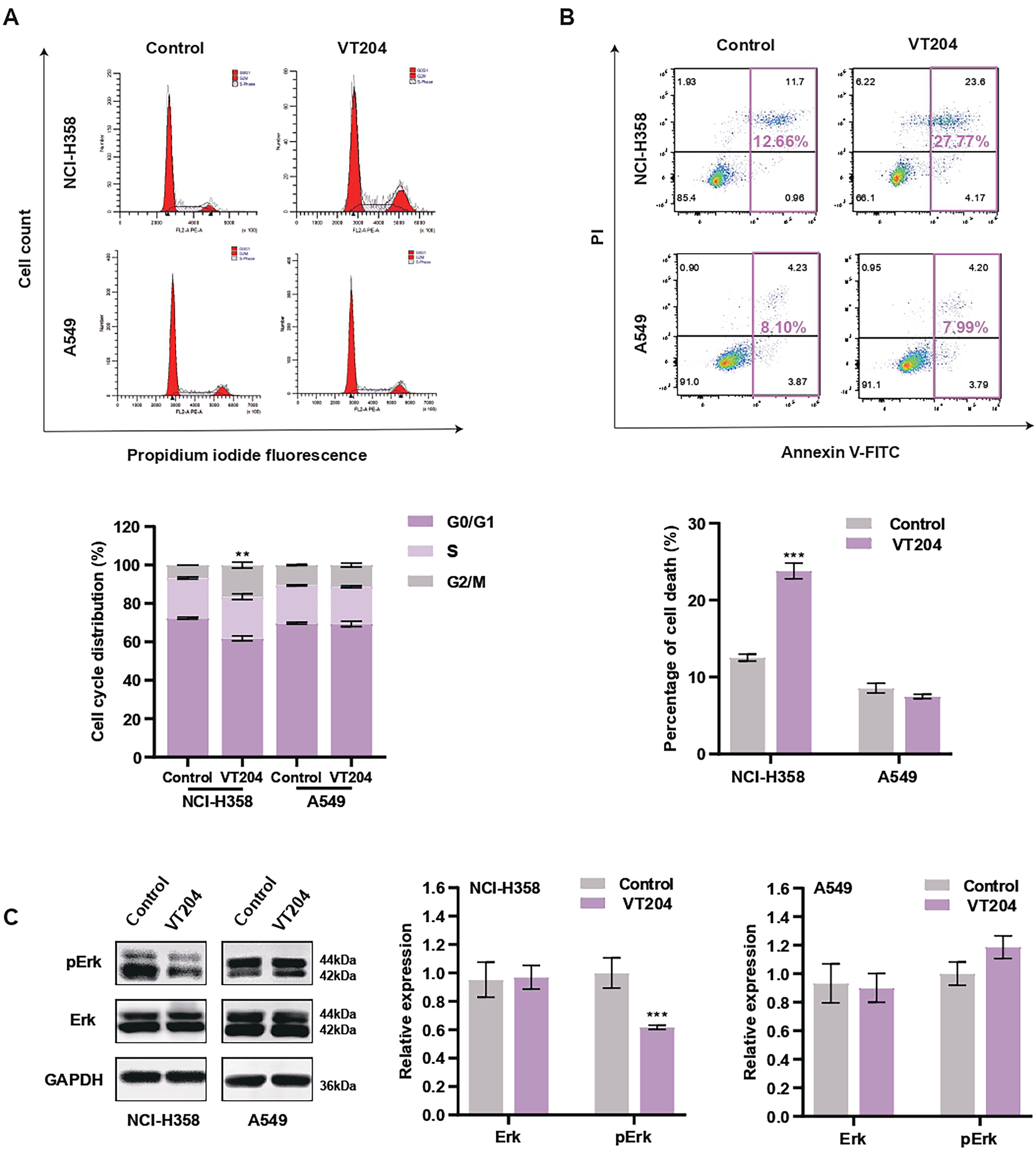
(A) Cell cycle distribution and quantitation in NCI-H358 and A549 cells treated with VT204 for 24 h in flow cytometry assays. (B) Representative results of Annexin V/FITC/PI staining by flow cytometry. (C) VT204 decreased pErk levels in NCI-H358 cells for 24 h. All the data shown represent results of 3 individual experiments (n = 3). Data were mean ± SEM. **P < .01, ***P < .001.

(A) In vivo efficacy evaluation of BALB/c-nude mice. (B) Representative images of NCI-H358 xenograft tumors in nude mice after 23 days of treatment with 50 mg/kg MRTX849 and VT204. (C) Tumor volume of NCI-H358 xenograft-bearing nude mice treated with MRTX849 and VT204. (D) Changes in body weight of nude mice inoculated with NCI-H358 during VT204 treatment. (E) Representative images of A549 xenograft tumors in nude mice after 23 days of treatment with 50 mg/kg VT204. (F) Tumor volume of A549 xenograft-bearing nude mice treated with VT204. (G) Changes in body weight of nude mice inoculated with A549 during VT204 treatment. Data were mean ± SEM. △P < .001 for all groups versus control, #P < .05 for VT204 versus MRTX849. (H) Effect of VT204 on the level of pErk in vivo. The data shown represent results of 3 individual experiments (n = 3). Data were mean ± SEM. ***P < .001. (I) Pathological analysis of lung, heart, liver, kidney, and spleen tissues after VT204 treatment. Scale bar = 1 mm.
In contrast, the administration of VT204 to A549 xenograft-bearing mice did not result in any significant difference in tumor volume compared to the control group (Figure 5E and F). These observations highlight that VT204 exhibits its pronounced tumor growth inhibitory effects in vivo primarily in KRASG12C-targeted NCI-H358 cells. Remarkably, the administration of VT204 did not cause any noticeable changes in the body weight of the treated mice, indicating that the drug was well tolerated and efficacious (Figure 5D and G).
Furthermore, our investigation aimed to explore the impact of VT204 on the KRAS downstream signaling pathway in vivo. Western blot analysis of tumor tissues revealed a significant reduction in pErk levels in nude mouse tumors injected with NCI-H358 cells in both the VT204 and MRTX849 groups (Figure 5H). Conversely, no significant difference in pErk levels was observed between the administered and control groups in nude mouse tumors injected with A549 cells. These in vivo results are in line with the outcomes obtained from in vitro cellular assays, supporting the specificity of VT204 in targeting KRASG12C.
To assess potential side effects of VT204 in vivo, various organs were collected and subjected to H&E staining. The results (Figure 5I) demonstrated no significant morphological abnormalities in the treated group compared to the control group. Collectively, our findings suggest that VT204 effectively inhibits tumor growth in NSCLC cells in vivo through its specific targeting of KRASG12C, while showing no evident impact on KRASG12S. These results highlight the promising potential of VT204 as a selective and well-tolerated therapeutic agent for KRASG12C-driven cancers.
Discussion
Despite significant efforts, the prognosis and overall survival of patients with NSCLC have not seen substantial improvement. 21 The complexity of KRAS signaling pathways and the crosstalk between these pathways present challenges and opportunities for targeted therapies and drug development.11,22,23 Covalent KRASG12C-binding small molecules such as AMG510 and MRTX849 have shown promise in overcoming NSCLC. 2 However, emerging clinical evidence suggests the development of resistance to KRASG12C inhibitors, necessitating the search for novel therapeutic approaches to effectively target KRASG12C and its resistance mechanisms. 11 Combination therapies have been explored to address this issue.22,24 In this study, we investigated the effects of VT204 on NSCLC A549 and NCI-H358 cells both in vitro and in vivo.
Our study aimed to uncover the precise targeting of VT204 on KRASG12C by examining its effects on 2 NSCLC cell lines. A detailed 3-dimensional structural comparison of KRASG12C and KRASG12S is important for elucidating the atomic-level mechanisms of VT204's selectivity. Conducting such a comparative study would not only highlight the structural nuances that confer VT204's specificity but also provide critical insights into the interaction dynamics at the molecular level. By analyzing the binding sites, we can identify key residues that differentiate the G12C mutation from the G12S mutation, enabling us to understand VT204's mechanism of action more thoroughly. This detailed analysis would indeed be pivotal for guiding the design of new inhibitors with enhanced selectivity, targeting specific conformational or compositional features unique to KRASG12C. We plan to incorporate these analyses into our ongoing research, providing a foundation for developing more refined and targeted therapeutic agents. The CCK8 experiments revealed that VT204 induced apoptosis in both NCI-H358 and A549 cells. Surprisingly, the inhibitory effect of VT204 on NCI-H358 cells was significantly greater, while the effective concentration on A549 cells was about 5-fold higher, suggesting that NCI-H358 cells are more sensitive to VT204. The A549 cell death observed at a concentration of 38 μM might be attributed to a toxic effect caused by the elevated DMSO ratio. Therefore, for subsequent experiments, a concentration of 8 μM, corresponding to the IC50, was selected after 24 h of treatment. This concentration effectively targeted the KRASG12C mutation in NCI-H358 cells but showed no significant difference in A549 cells compared to the control group. The CCK8 assay demonstrated a concentration-dependent decrease in the survival rate of NCI-H358 cells after incubation with VT204. Two possible underlying mechanisms for this effect are induction of cell autophagy, apoptosis, necrosis, or other cell death pathways, and interference with the cell cycle to hinder cell proliferation.3,20,25 To investigate how VT204 inhibits the survival of NCI-H358 cells and its effect on the cell cycle, flow cytometry was used to analyze cell cycle distribution and apoptosis in NCI-H358 and A549 cells. Similar analyses were performed for A549 cells as well.
Our analysis of the cell cycle distribution revealed differences in the response to VT204 treatment between NCI-H358 and A549 cells. VT204 treatment of NCI-H358 cells resulted in a significant increase in the proportion of cells in the G2/M-phase, indicating G2/M-phase arrest. However, G2/M-phase arrest was not observed in A549 cells treated with VT204. Cell cycle progression is an important process that supports cell proliferation, and this process is regulated by cell cycle-related proteins.20,26 These findings suggest that VT204 effectively arrests KRASG12C-mutated cells in the G2/M-phase, which might be attributed to the downregulation of cyclin B1 and cyclin-dependent kinase 1 expression.27,28 As cell cycle progression was blocked at the G2/M-phase, cells may subsequently undergo apoptosis, which is one of the key mechanisms by which cancer therapy induces cancer cell death. 20 Furthermore, apoptosis induction by VT204 was not observed in KRASG12S-mutated A549 cells. These results collectively suggest that VT204 exerts cell cycle-specific effects on NCI-H358 cells and exhibits apoptotic characteristics. Additionally, our study demonstrated that VT204 significantly inhibited the migration and invasive abilities of NCI-H358 cells. In light of previous studies and our findings, VT204 shows promise as a therapeutic agent targeting KRASG12C in NSCLC.
Recent studies have identified multiple signaling pathways and specific regulatory molecules associated with the anti-tumor mechanisms of KRASG12C-targeted therapeutic agents. Similar to other KRASG12C inhibitors, this class of inhibitors specifically targets KRASG12C and does not affect RAS signaling in non-malignant cells, minimizing the risk of on-target, off-tumor toxicities.29,30 VT204, like other clinical KRASG12C inhibitors such as sotorasib and adagrasib, acts by inhibiting KRASG12C in the GDP-bound state. 6 In vitro Western blotting analysis showed that VT204 treatment reduced pErk levels in NCI-H358 cells, indicating inhibition of KRAS activity, while no significant difference was observed in A549 cells. It has been established that activating KRAS leads to ERK phosphorylation, and the reduction of pERK levels proves that KRAS is inhibited. 31 Therefore, pERK was used as an indicator of KRAS activity in this study. In vivo experiments using xenograft models showed that VT204 exhibited potent tumor suppressive effects comparable to MRTX849 (adagrasib), another highly selective KRASG12C inhibitor. Western blot analysis of tumor tissue confirmed that VT204 reduced pErk levels in vivo, consistent with the in vitro findings. These results align with the previously reported mechanisms of action of KRASG12C inhibitors. 31 Molecular docking studies revealed that VT204 had a docking score similar to that of MRTX849 when binding to the KRASG12C protein at the switch-II pocket. Previous studies have identified MRTX849 as an irreversible covalent inhibitor of KRASG12C that forms a covalent bond with cysteine 12. 32 RAS functions as a binary molecular switch, cycling between the active state (GTP-bound) and the inactive state (GDP-bound). 33 Constitutive activation of mutated Ras can activate the MAPK (Ras-Raf-Mek-Erk) signaling pathway and ultimately lead to cancer development. 34
Furthermore, the in vivo antitumor efficacy of VT204 was evaluated in comparison to MRTX849, serving as a control to better observe the pharmacological effects of VT204. Nude mice injected with NCI-H358 cells were administered VT204, MRTX849, or a control treatment. After 2 weeks of alternate-day administration, significant tumor volume reduction was observed in the administered groups. Treatment was then discontinued to facilitate subsequent experiments, and tumor volume was monitored until day 23. The results showed a significant decrease in tumor volume during the first 2 weeks compared to the control group, with similar tumor inhibition rates observed in the VT204 and MRTX849 groups, indicating the potent inhibitory effect of VT204 on KRASG12C-induced tumor growth. However, no significant change in tumor volume was observed in nude mice injected with A549 cells after administration of VT204, suggesting that VT204 was ineffective against KRASG12S targets. This is the first in vivo nude mouse experiment with VT204, with reference to the positive drug MRTX849 showing dose-dependent antitumor efficacy in a well-tolerated dose range, with a maximum effective dose between 30 and 100 mg/kg per day. 5 A moderately effective dose of 50 mg/kg was selected, and in order to see the difference in in vitro inhibitory effect between VT204 and MRTX849 at the same concentration, the same 50 mg/kg was chosen.
In summary, our study demonstrated the inhibition of cell proliferation by VT204, which was associated with comprehensive and sustained inhibition of KRAS-dependent signaling and the induction of apoptotic responses. These findings highlight the potential of VT204 as a therapeutic agent for NSCLC targeting KRASG12C. Elucidating the mechanisms underlying the therapeutic response to KRAS inhibition by VT204 provides insights into strategies to enhance the therapeutic activity against KRAS-mutant tumors. The discovery of VT204 supports the use of covalent KRASG12C inhibitors for the treatment of KRASG12C-mutated cancers and provides a clear direction for further investigation. In conclusion, our results indicate that VT204 is a potent and selective KRASG12C covalent inhibitor with promising potential for NSCLC therapy.
While the study presents significant findings regarding the therapeutic potential of VT204 in targeting the KRASG12C mutation in NSCLC, the study's experimental design primarily focuses on in vitro and in vivo responses in a single cell line model, which might not fully represent the complexity of human tumors. The use of a broader range of KRAS-mutated cell lines could provide a more comprehensive evaluation of VT204's efficacy and specificity in future.
Conclusions
In conclusion, our study provides compelling evidence that VT204 exerts a potent inhibitory effect on the proliferation of NCI-H358 cells. Through its intervention in Erk phosphorylation, VT204 effectively induces apoptosis in these cells. These findings suggest that VT204 holds promise as a KRASG12C inhibitor for the treatment of NSCLC. Further studies are warranted to elucidate the precise molecular mechanisms underlying the therapeutic effects of VT204 and to optimize its efficacy in clinical settings. The discovery of VT204 as an effective and selective KRASG12C covalent inhibitor adds to the growing body of evidence supporting the use of targeted therapies against KRAS-mutated cancers.
Supplemental Material
sj-docx-1-tct-10.1177_15330338241264853 - Supplemental material for VT204: A Potential Small Molecule Inhibitor Targeting KRASG12C Mutation for Therapeutic Intervention in Non-Small Cell Lung Cancer
Supplemental material, sj-docx-1-tct-10.1177_15330338241264853 for VT204: A Potential Small Molecule Inhibitor Targeting KRASG12C Mutation for Therapeutic Intervention in Non-Small Cell Lung Cancer by Xuechao Yang, Shu Zhang, Yang Yang, Xiaoqun Duan and Xiaochuan Li in Technology in Cancer Research & Treatment
Footnotes
Abbreviations
Author Contributions
XY: conceptualization, data curation, formal analysis, methodology, visualization, writing—original draft, writing—review and editing. SZ: conceptualization, data curation, methodology. YY: validation, writing —review and editing, supervision. XD: project administration, funding acquisition, resources. XL: writing—review and editing, funding acquisition, resources
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Major Science and Technology Project of Guangxi province, China, grant number “AA22096025” and the Innovation Project of Guangxi Graduate Education, grant number “YCSW2023431”.
Ethical Approval and Consent to Participate
This study followed the Guide for the Care and Use of Laboratory Animals, all animal experiments were approved by Animal Care and Use Committee of Guilin Medical University (the approval number is GLMC-IACUC-2023004). Manuscript approval date is August 3, 2023.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.