
Abstract
Epigenetic machinery is a cornerstone in normal cell development, orchestrating tissue-specific gene expression in mammalian cells. Aberrations in this intricate landscape drive substantial changes in gene function, emerging as a linchpin in cancer etiology and progression. While cancer was conventionally perceived as solely a genetic disorder, its contemporary definition encompasses genetic alterations intertwined with disruptive epigenetic abnormalities. This review explores the profound impact of DNA methylation, histone modifications, and noncoding RNAs on fundamental cellular processes. When these pivotal epigenetic mechanisms undergo disruption, they intricately guide the acquisition of the 6 hallmark characteristics of cancer within seemingly normal cells. Leveraging the latest advancements in decoding these epigenetic intricacies holds immense promise, heralding a new era in developing targeted and more efficacious treatment modalities against cancers driven by aberrant epigenetic modifications.
Introduction
Genetic code or information within DNA sequence is transformed into protein expression through the central dogma, influenced by genetic and environmental factors that regulate genes via multiple chemical interactions. Central dogma, through which genetic code or DNA is converted into protein expression, depends on genetic and environmental factors that control genes through several chemical interactions. The eukaryotes’ genome is modulated by epigenetic changes that lead to aging or cancer progression through modulating DNA replication and repair processes, regulating genomic instability and X-chromosome imprinting, and establishing transcription profiles. 1 The epigenetic modifications such as histone alterations and DNA methylation complexly control gene expression, affecting biological functions beyond simple genetic coding. They are necessary for the growth and development of embryos, cell differentiation, and responses to external stimuli. Through modifications to the typical gene expression patterns, dysregulation of these epigenetic pathways can be a key contributor to the advancement of various illnesses, like cancer. By understanding these complex alterations, illnesses associated with epigenetic dysregulation may provide new targets for treatment, opportunities for precision medicine, and tailored therapeutics. 2
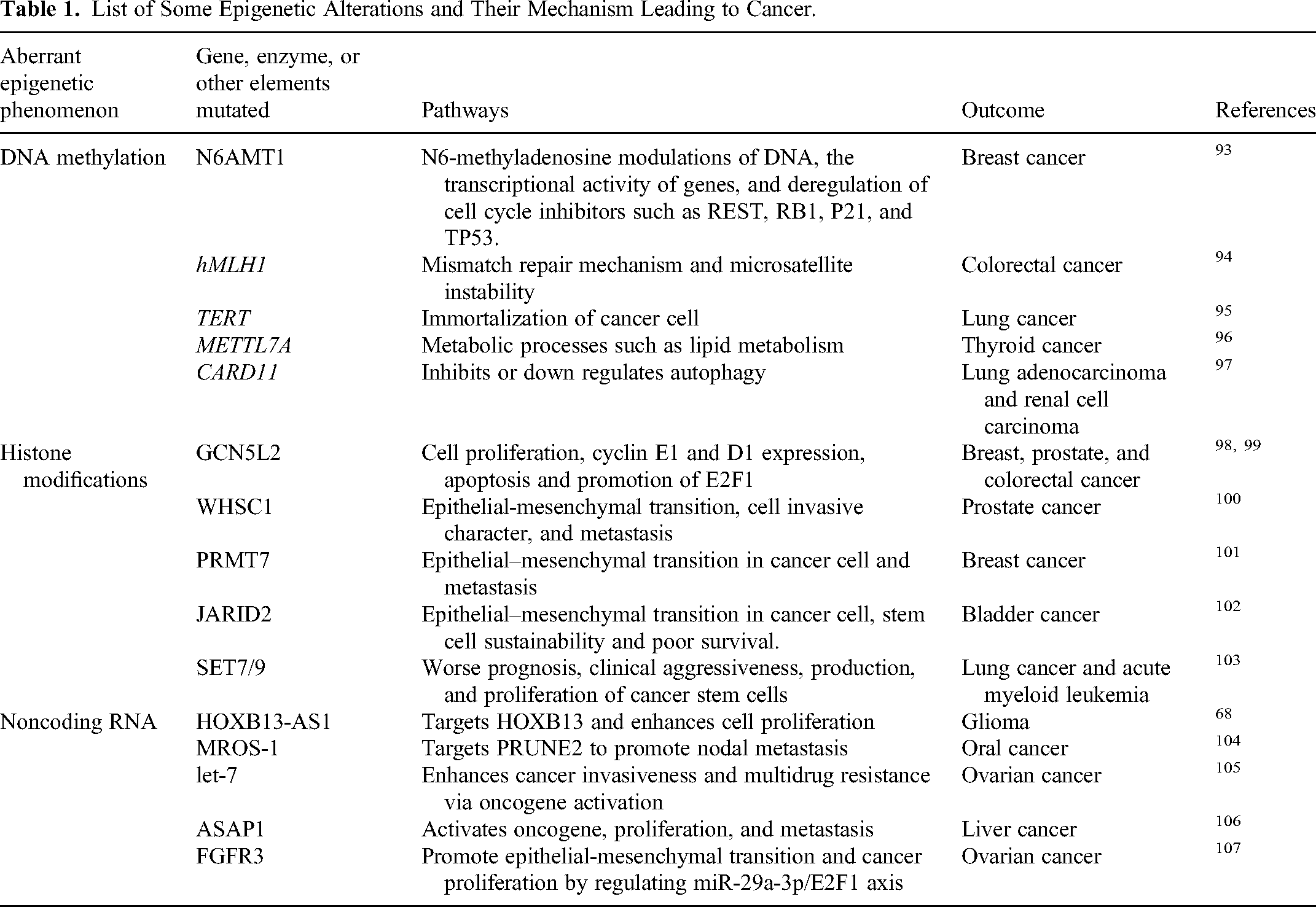
Cancer is the most severe and prevalent cause of mortality, characterized by uncontrolled cell proliferation, with a mortality rate of 10 million deaths worldwide up till 2020. 3 Cancer is considered a genetic disease; however, epigenetic changes that regulate gene expression are the key contributor to adult and embryonic life, and their deregulation can cause major diseases, including cancer. 4 Hanahan and Weinberg proposed 6 hallmarks of cancer, including sustained proliferative signaling, resistance to cell death, avoiding growth suppressors, stimulation of angiogenesis, active metastasis and invasion, and enabling replicative immortality. 5 The genetic reasons behind cancer include the activation and upregulation of oncogenes and the inactivation or downregulation of tumor suppressor genes due to mutations. 6 Oncogene mutations cause hyperactivation, which encourages unrestrained cell growth and proliferation. In contrast, tumor suppressor gene mutations cause loss of function, which permits unrestricted cell division and inhibits systems that ordinarily prevent tumor development. During carcinogenesis, one of the epigenetic alterations, DNA methylation, a dynamic activity, occurs in 2 forms: first, the demethylation of oncogene promoters, and second, the de novo methylation of selected CpG islands in tumor suppressor genes to inactive them. 7 In order to design tailored treatment plans and uncover targeted medicines for cancer, it is essential to comprehend these genetic abnormalities. 8
Epigenetics is defined as a heritable modification in chromosome structure and stability or gene expression through DNA methylation, noncoding RNAs, or histone covalent modifications without altering DNA sequence. 9 The term “epigenetics” was described in 1942 by Waddington. Waddington described epigenetics as the development of phenotype due to external and internal interaction between genes and the environment. Epigenetic machinery consists of DNA coiled around histone proteins in a nucleosome. 1 Major epigenetic regulatory mechanisms involve DNA methylation catalyzed by DNA methyl transferases, histone methyl transferases (HMTs), and histone demethylases regulate histone methylation, histone acetyltransferases, and micro-RNase activates histone acetylation catalyze RNA silencing in normal cells. 10 These 3 significant epigenetic changes act alone or intermingle with each other to regulate cancer progression. For example, the methylation of DNA and histone affects the transcription of target genes. DNA methylation or histone modifications can also promote or inhibit alternative splicing to enhance the diversity of noncoding RNAs involved in cancer metastasis. Histone modifications or DNA methylation have a direct effect on alternative splicing for example aggregation of the H3K36me3 (histone H3 lysine 36 trimethyl) signals on exons linked with lower levels in alternatively spliced exons compared to constitutive exons. Several studies also showed local DNA methylation influences the splicing patterns of individual cassette exons during cell differentiation. 11 In a study, Flavahan et al explained how misregulation in any of these epigenetic mechanisms can contribute to the hallmarks of cancer by raising oncogenic properties in normal cells and converting them into tumor cells. 12 Epigenetic machinery mutations are the predominant cause of human cancers. 1
Cancer-causing genetic alterations are quite challenging to reverse; however, epigenetic changes can be pharmaceutically reversed. 13 The relationship between tumorigenesis and epigenetics is highly intertwined through the proliferation, metastasis, and heterogeneity of cancer cells. 14 Epigenetic alterations are represented as malignant tumor formation in cancer. 10 Chemotherapy is considered a traditional method to treat advanced cancers. After chemotherapy, abrupt epigenetic changes can be caused by chemotherapeutic agents, which cause chemoresistance, serving as an obstacle to cancer therapy. This requires additional research into alternative therapeutic approaches. 4 Both epigenetic drugs that target epigenetic mechanisms involved in tumor initiation and utilization of emerging epigenetic tools can be used as a practical approach to eradicating various human diseases. 13 These epigenetic drugs include inhibitors of DNA methyl transferases (DNMTi) and histone deacetylases (HDAC) 15 and nucleoside analogs (decitabine and azacytidine) approved by the FDA. 16 This review article summarizes epigenetic changes in DNA methylome, histone modification, and miRNAs in cancer, the contribution of epigenetics in gene expression, epigenetic alterations, and biomarkers in different types of cancer, and targeting epigenetics for cancer therapy. This article mainly focuses on the new and improved methods for early detection and prevention of cancer, identifying new prognostic markers for cancer based on epigenetic changes, and using reversibility of epigenetic modifications for drug development.
Epigenetic Basics
Epigenetics is a reversible process that affects gene expression. Epigenetic mechanisms in normal mammalian cells are important in regulating and maintaining tissue-specific expression of genes, stabilizing chromatin structure, and controlling normal development. 17 Due to defective epigenetic mechanisms, tumorigenesis occurs, accumulating 6 hallmarks of cancer in normal cells. Epigenetic modifications are well balanced in normal cells for developmental processes like genomic imprinting, X-chromosome inactivation in females, changes in cytosine-methylation patterns of DNA, and the nucleosome positioning along DNA and maintenance of chromatin structure. Evidence suggests that plenty of genetic and environmental conditions disrupt pathologies, including cancer. 1
DNA Methylation
The process of DNA methylation includes the accretion of methyl group to the fifth carbon atom of cytosine present in the CpG cluster. This epigenetic mark is catalyzed by DNA methyltransferases (DMTs) and affiliated with heterochromatin condensation and gene repression. The most common target of DNA methylation, CpG islands, is typically present at the 5’ end of several genes. 18 In mammals, 80% of CpG islands are methylated, but in normal cells, the region within 2 kb of the islands is known as CpG shores, and some CpG islands are exceptionally hypomethylated. In the mammalian genome, 85% to 100% of regions are methylated, while 0 to 5% are unmethylated. In mammals, 80% of CpG islands are methylated. DNMT3-A and DNMT3-B catalyze the de novo methylation of double-stranded DNA, whereas DNMT1 maintains the DNA methylation pattern of newly synthesized strands. 18 DNMT3-A and DNMT3-B belong to a protein family DNMT3 that consists of an enzyme domain at the C-terminal region, an ATRX-DNMT3A-DNMT3L (ADD) domain in the central region, and Pro-Trp-Trp-Pro (PWWP) domain at the N terminal region. The regulation of enzymatic activity and the localization of DNMT3A and DNMT3B to their target sites is performed by both the PWWP and ADD domains. The PWWP domain (reader module) is involved in the proper chromatin binding of DNMT3A and DNMT3B and in recognizing histones H3K36me2 and H3K36me3, while the ADD domain (molecular module) recognizes unmodified histone H3 lysine 4. 19 DNA C5-cytosine methylation (5mC) and RNA N6-adenosine methylation (m6A) are predominant methylation mechanisms; however, apart from these, N6-methyldeoxyadenine is prevalent in both prokaryotes and eukaryotes mediated by 6mA MTases (methyl transferases). 20 In tumor suppressor genes, gene silencing resulting from DNA methylation leads to carcinogenesis. In the mammalian genome, 85% to 100% of regions are methylated, while 0 to 5% are unmethylated. In housekeeping genes, the CpG island promoters are rarely methylated, while this modification occurs in those gene promoters or enhancers that are stabilized in a silenced state. 1 DNA methylation of repeat regions maintains genome integrity by preventing retrotransposon activity under normal physiological conditions. In females, it helps in the deactivation of one of the copies of the X-X-chromosome and suppressing genes of specific tissues. 21 A prime example of a DNA methylation mechanism in normal cells is the activity of the ubiquitous transcription factor, Specificity protein 1 (Sp1). This protein interaction with DNA is controlled by the presence or absence of DNA methylation at CpG islands. Sp1 protein associates with unmethylated DNA to promote gene transcription while binding to methylated CpG sites is inhibited, therefore promoting gene transcriptional silencing in somatic cells. Altered DNA methylation patterns in cancer metastasis consist of a combination of distal enhancers and promoters overlapping transcriptional start sites hypermethylation causing critical gene regulatory elements repression associated with hypomethylation of retro-elements, centromeres, and oncogenes. 22 For example, the deamination of 5-methylcytosine to thymine causes a mutation in the tumor suppressor gene, or proto-oncogene produces an oncogenic point mutation that disrupts standard cellular mechanisms, contributing to cancer metastasis. 23
Histone Modifications
Histone proteins posttranslational modification are associated with chromatin structure. A nucleosome comprises 147 bp DNA molecule wrapped around a histone octamer composed of 2 copies of 4 histone proteins, H2A, H2B, H3, and H4. All nucleosomes combine through linker DNA and histone H1 to form chromatin. 24 Histone proteins are rich in positive amino acids lysine and arginine. Histone proteins are a part of nucleosomes rich in positive amino acids lysine and arginine. The most prominent histone modifications are phosphorylation, acetylation, ubiquitination, ADP ribosylation, sumoylation, 25 and methylation, resulting in chromosome remodeling. Histone, a prominent part of chromatin, consists of globular domains and tails in which long tails are more susceptible to these covalent modifications that further modulate chromatin structure and gene expression. 26 In histone modification, specific residues are important in maintaining gene expression and genome integrity and avoiding cancer risk. Histone core proteins and N-terminal amino acid tails are hotspots for their posttranslational modifications. A ubiquitination site is represented at histone 2 (2A and 2B), methylation sites at histone 3 and 4 (H3K9, H3K4, H3K36, H3K27, H3K79, and H4K20), phosphorylation areas at H3S10 and acetylation of amino acids including H3K23, H3K18, H3K9, H3K14, H4K5, H4K12, H4K8, and H4K16.
All these modifications play a vital role in cell DNA replication, DNA repair, chromosome dynamics, and DNA condensation during mitosis and meiosis. They are involved in the interaction of histones with chaperones, proteins, and DNA, etc. In the methylation process, arginine residue is modified by 1 or 2 methyl groups that can be arranged symmetrically or asymmetrically. At the same time, lysine can also contain 1, 2, or 3 methyl groups that do not affect the positive charge of lysine residue. 24 Deregulation of HMTs and histone methyl demethylases (HDMs) can cause breast, prostate, lung, and brain cancer. 1 Acetylation modification is carried out by histone acetyltransferases (HATs) and HDAC. Acetylation plays a prominent role in molecule charge and DNA unwinding processes. A single acetyl group can bind in the middle of a lysine residue, affecting its positive charge. Histone PTM comprises the addition or removal of small groups: acetylation (acetyl group in lysine residue), methylation (methyl in lysine, arginine, and glutamine residues), phosphorylation (phosphoryl groups in serine and threonine residues), or entire protein in the case of ubiquitin or small ubiquitin-like modification (SUMO) in lysine residue by specific enzymes. 24
Noncoding RNAs
Approximately 75% of the human genome is transcribed into RNA, out of which only 3% encodes for proteins while 72% is not translated to proteins referred to as noncoding RNA. 27 On the basis of length, shape, and location, there are 4 significant types of noncoding RNA: microRNA (miRNA), long ncRNA (lncRNA), circular RNA (circRNA), and PIWI-interacting RNA (piRNA) that function distinctly in cancer. MicroRNA is composed of 22 nucleotides and are small RNA that cause RNA-induced silencing complex by binding the corresponding sequence in targeted mRNA to degrade it. 28 PiRNA is 24 to 30 nucleotides long in length, first identified in Drosophila, present in germ line cells that mediate in epigenetic regulation of chromatin 29 while both lncRNAs and circRNAs are >200 nucleotides long; however, lncRNAs are linear and circRNAs are ring-like. 30 When transcribed, both can fold into secondary structures and enable their interactions with DNA, RNA, and proteins. LncRNAs and circRNAs are involved in the epigenetic modulation of chromatin to regulate gene expression and act as a scaffold to regulate signaling pathways and protein–protein interactions. At transcriptional and posttranscriptional levels, ncRNAs contribute to the regulation and activity of cyclins, cyclin-dependent kinases, and their inhibitors mainly involved in the cell cycle in normal cells. 31 For instance, expression of lncRNA is enhanced in response to DNA damage; however, expression of microRNA is modulated by cell-cycle-dependent transcription factors such interactive patterns, when altered, can initiate and progress cancer by acting as an oncogene or suppressor leading to human diseases. 20 These ncRNAs can help to initiate and progress cancer by acting as an oncogene or suppressor leading to human diseases. 32
Epigenetic Control of Gene Expression
Different epigenetic modifications, including genomic DNA methylation, histone modifications, and microRNA regulation, contribute toward the normal development of cells, thus maintaining cell identity. Epigenetic factors assure cell-type specificity in normal development. Epigenetic modifications decrease the expression of pluripotency-related genes while enhancing the expression of housekeeping genes and appropriate patterns of tissue-specific genes during normal development. 33
Epigenetic Control of Normal Cellular Processes
In somatic differentiated cells, 2 critical epigenetic modifications, DNA methylation and histone modifications allow the expression of tissue-specific and housekeeping genes to control cell differentiation only (Figure 1). DNA methylation plays a crucial role in X-inactivation, genomic stability, differentiation, and imprinting. 34 A number of genes are methylated in somatic cells but unmethylated in germ-line cells. DNA hyper-methylation of repetitive genomic sequences serves as a mechanism to prevent chromosomal instability, translocations, and gene disruption caused by transposons. It has been suggested that some transposon families are both methylated and modified by histone proteins. To equalize the imbalance of the “extra” X-chromosome gene expression as compared to the one X-chromosome in males, DNA methylation is known to lead to X-chromosome inactivation, which causes monoallelic gene repression in female cells. 35 To study the relationship between histone modifications and developmental expression of genes, the β-globin locus is the best example, as at different stages of development, vertebrates express different globin genes. 36 MicroRNAs (miRNAs) are involved in cellular phenomena such as proliferation, development, differentiation, and apoptosis. They are tissue-specific in their expression. For instance, brain-specific miR-184, an intrinsic modulator of adult neurogenesis, is imprinted and subjected to epigenetic control by methyl-binding protein MeCP2 release. 33

Epigenetic phenomenon in normal mammalian cells. These processes control and tune chromatin structure as well as gene expression. (A) DNA methylation, (B) histone modification, and (C) noncoding RNAs.
Epigenetic Remodeling Leading to Cancer
Altered DNA Methylation Patterns in Cancer
Initiation and advancement of cancer are coexistent with a broad range of epigenetic aberrations; such as changes in basic DNA methylation patterns. These changes can be hypomethylation in repetitive DNA sections, laminin-associated domains, and retrotransposons with lesser CpG sites (Figure 2). Hypomethylation ultimately leads to switching on the transposable elements and genetic instability. 37 Feinberg and Vogelstein first discovered this aberrant methylation changes with a global decline in 5mC content among various cancer types. 38 The higher aneuploidies and mutation rates that resulted from the reduction or deletion of DNA methyl-transferase (Dnmt1) have highlighted the contribution of DNA hypomethylation to induce chromosomal instability and fragility.39,40 The reduction in methylated DNA activates the transcription of oncogenes 41 or the regions with transposons, consequently enhancing chromosomal recombination and genome instability. 42 The frequent hypomethylation of transposable elements enriched in intragenic and intergenic fragments accelerates gene expression. For instance, a study demonstrated how the oncogene is turned on due to DNA hypomethylation of CpG sites within the L1NE1 promoter and the consequent generation of favorable chromatin structure at the MET promoter. 43 Likewise, hypomethylation of SINE repeats is detected in victims of acute myeloid leukemia. 44 The hypomethylation of IGF2, PLAU, CDH3, BCSG1, and FEN1 is detected to be a prominent alteration in breast cancer patients. 45 The reduction of methylation in pericentromeric areas of chromosome number 1 or 16 is frequently noted in a variety of carcinomas. 40 Studies conducted on metastatic tumors of nonsmall lung cancer patients elucidated the upregulation of cell motility 3 and putative-oncogene engulfment elements due to hypomethylation within their promoter region. 41 Moreover, promoter hypomethylation is correlated with gain in the expression of Iroquois homeobox-1 in metastatic osteosarcoma. 46
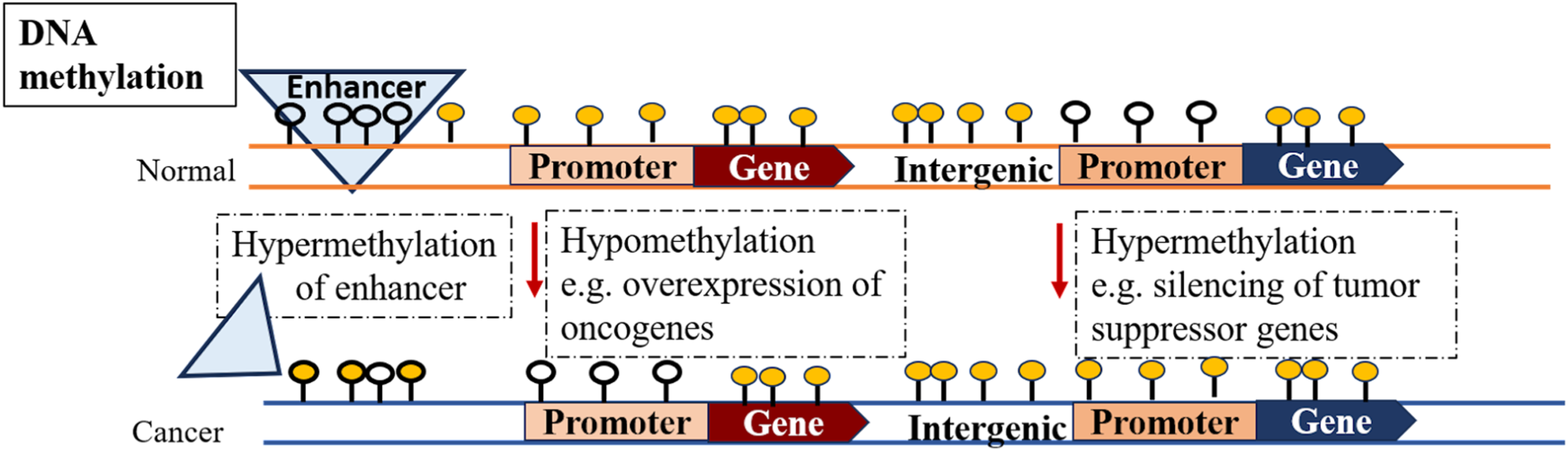
Comparison of DNA methylome in typical and cancer cells.
The aberrant hypermethylation in 5’ CpG sites of the gene is another central alternated methylation pattern resulting in cancer progression. Usually, CpG islands (CPI) located in 60% of gene promoters are nonmethylated. The deficiency of DNA methylation is accountable to the activated gene expression in open chromatin regions. 47 Studies on various types of cancer cells showed that hypermethylation in CPI of genes deactivates expression of tumor-suppressor (Figure 2), DNA repair, and cell cycle regulatory genes.48–50 Research on colorectal cancer and gliomas has confirmed the promoter's hypermethylation of a DNA repair enzyme named O6-methyl-guanine methyltransferase. Inactivation of this enzyme intern makes KRAS and p53 genes vulnerable to genetic mutations.51,52
Similarly, malignant renal cell carcinoma is initiated due to the silencing of the VHL tumor such oppressor through the promoter's hypermethylation. 53 The absence of imprinting at the 1GF2/H19 locus due to abnormal methylation is common in Wilms’ tumor, which in turn contributes to activation and advancement of gastric and colorectal cancer due to overexpressed growth factor IGF2. 54 Abnormal hypermethylation of a RASSF4 (involved in negative regulation of RAS pathway) and GPX3 genes (stabilizes reactive oxygen species to suppress growth) lead to the development of multiple myeloma.55,56 In myelofibrosis patients, silencing of a transcriptional regulator ZFP36L1 (a tumor suppressor gene) takes place owing to aberrant methylation of its enhancer, and deregulation of the gene is responsible for neoplastic transformation. 57
Remodeling of methylation landscape is possibly accompanied by mutations in 10-11 translocator protein-2, a methyl-cytosine dioxygenase that oxidizes 5-methylcytosine (5mC) into 5- hydroxy-methyl-cytosine (5hmC), its successive conversion into 5-formylcytosine and 5-carboxymethylcysteine.58,59 The consequent excision and replacement of 5-carboxymethyl cytosine with cytosine to thymine DNA glycosylase contribute to methylation alteration. 60 Impaired hydroxylation is observed in almost 15% of myeloid cancers due to mutations in TET2 or functional losses in DMTs. 61
It has been demonstrated through previous studies that chromosome remodeling, transcription factors, 62 histone modification, 63 and lncRNA 64 play significant roles in regulating DNA methylation in various carcinomas. It has been conclusively demonstrated through studies on colon and esophageal cancer that lncRNA-mediated recruitment of DNMT1 65 in cancer cells and direct recruitment of DNMTs at the promoter regions through LINC01270/HOTAIR, respectively, are the predominant mechanisms.66,67 Whereas indirect recruitment of DNMTs through the association of lncRNA with enhancer of zest-homolog-2 is a common phenomenon in several carcinomas. 68 Another study conducted on digestive cancer patients showed the contribution of lysine methyl transferases, protein-arginine methyl transferases, and lysine-demethylases; transformers of histone methylation; in metabolic reprograming, carcinogenesis, invasion, and epithelial–mesenchymal transition of digestive tissues 69 thus provided new aspects as therapeutic agents to administer such cancer.
Aberrant Histone Modification
Covalent modifications of histones, such as phosphorylation, acetylation, and methylation, are involved in reshaping the genome, nucleosome dynamics, chromatin compaction, and transcription of genes 70 ; thus, their aberrant patterns are frequent in several types of cancers (Figure 3). These modifications are controlled by HMTs, HATs, HDMs, and HDACs. 71 Under normal conditions, histones located at enhancers and promoters of activated genes were found to be enriched by H3K27 acetylation. The genome-wide analysis confirmed the accumulation of aberrant H3K27 acetylation, which activates tumorigenic enhancers, thus resulting in the advancement of cancer. 72 Aberrant histone methylation, such as H3K27me3 or H3K4me3, is proposed to enhance the adaptation and plasticity of the tumors to their surroundings. 73 Colorectal, prostate, and hepatocellular cancer progression occurs due to the upregulation of H3K4 HMT encoded by SMYD3 74 and H3K4 HMT encoded by EZH2 75 . Mutations of KMD5C, KMD5A (influencing methylation of H3K4), and KMD6A (influencing methylation of H3K27) are prevalent in several carcinomas. 76 Studies on hematological cancer have confirmed CREBBP and EP300 chromosomal translocation in genes encoding HATs. 77 Similarly, EP300 chromosomal translocation is commonly observed in breast, colorectal, pancreatic, and gastric tumors. 78
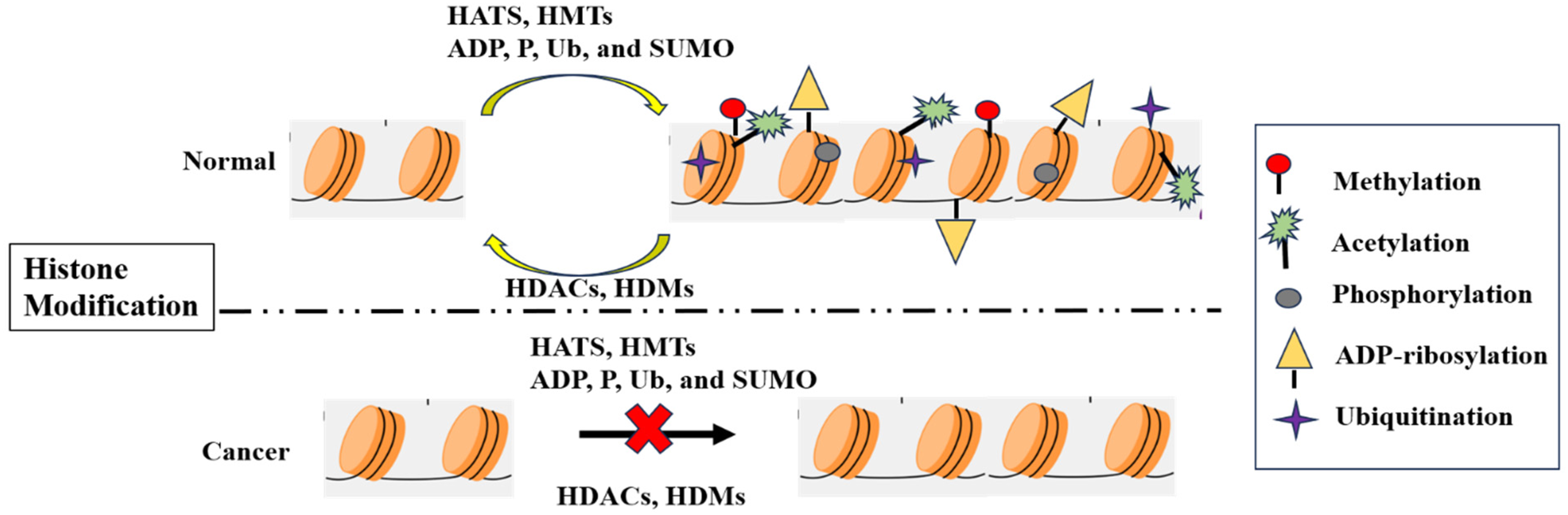
A comparison of general histone modifications in normal and cancer cells.
Histone phosphorylation controls several phenomena, namely DNA repair, mitosis, and gene regulation, and aberrant phosphorylation patterns of histones are frequently detected in prostate, colorectal, and breast cancer. 79 Interestingly, overexpressed protein phosphatase 2A is diagnosed as a risk factor for the progression of hepatocellular cancer in patients with chronic hepatitis. 80 Correspondingly, the advancement of hepatocellular carcinoma is enhanced due to a decrease in histone-H2A ubiquitination. 81 The overexpression of a SUMO conjugated enzyme Ubc9 is customarily accompanied by the expansion of myeloma, lung cancer, ovarian cancer, 82 and hepatocellular carcinoma. At the same time, its downregulation enhances levels of cleaved-Caspase-3. 83 Similarly, inhibiting ADP-ribose polymerases binding to Sirt6 by a viral protein induces hepatocellular carcinoma. 84
Noncoding RNAs and Cancer
Being the regulators of genes, alteration in noncoding RNAs (ncRNAs), including deletions and insertions, are associated with the initiation and advancement of several carcinomas. A study on chronic lymphocytic leukemia showed that a 13q14 deletion in genes encoding miR-15 and miR-16 RNAs is responsible for the progression of the disease. 85 Inactivation of microRNAs through promoter hypermethylation is often detected in cancer. For example, oncogenic genes comprising CDK6, E2F3, C-MYC, and TGIF2 are activated due to silencing regulatory miR-9-3, miR-148, and miR-34b/c via methylation. 86 Proteins and peptides such as FBXW7-185aa, SHPRH-146aa, HOXB-AS3, and PINT-87aa encoded by ncRNAs regulate epithelial-to-mesenchymal transition, glucose metabolism, and ubiquitination pathway lead to tumorigenesis. 87 An m6A mutation in various circular RNAs is found to be responsible for initiating a variety of cancers, such as circ-NSUN2, modified by m6A, which establishes colorectal cancer, 88 and circE7, modified by m6A, which establishes cervical cancer. 89
Similarly, modifications in another class of ncRNAs called long-ncRNAs are prevalent in several carcinomas. For example, an m6A-altered LINC00958 (IncRNA) interns upregulate HDGF and are thus involved in the progression and lipogenesis of hepatocellular cancer. 90 Moreover, research on HCC patients showed a miR-29a mediated 5hmC modification that promotes tumorigenesis via the TET-SOCS1-MMP9 pathway.91, 92 Figure 4 shows a general mechanism by which ncRNAs induce oncogenesis. In a nutshell, some epigenetic modifications and their mechanism leading to cancer are demonstrated in Table 1.
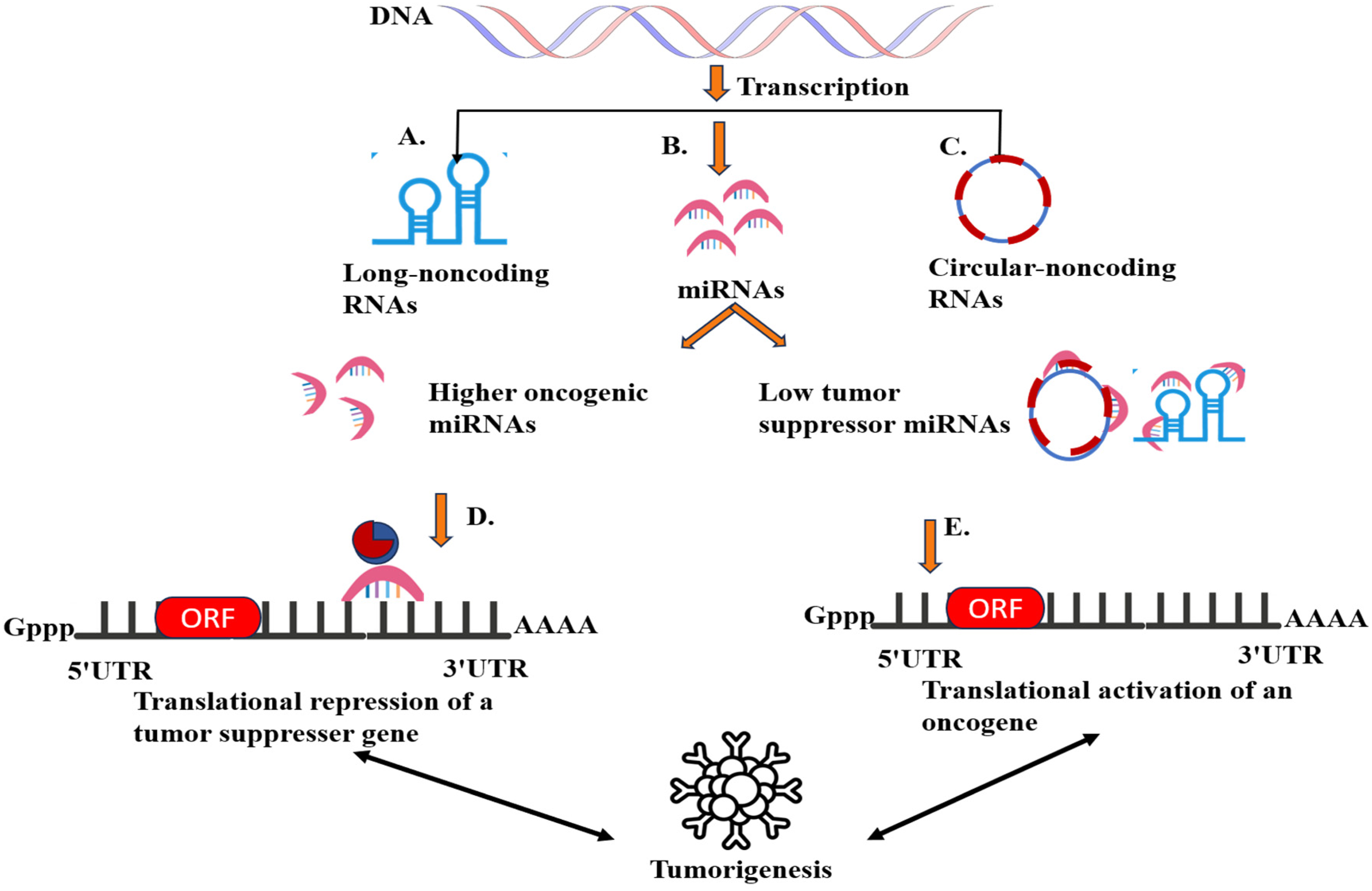
Contribution of ncRNAs in oncogenesis. (A) IncRNAs can either interact with the promotor or repressor and activate or silence a gene, interact with the chromatin-remodeling complex, and sequester miRNAs to inhibit their binding on mRNA. Deregulation of IncRNAs intern represses tumor suppressor genes or upregulates oncogenic signaling. 91 (B) miRNAs modulate genome expression by binding with mRNA. The silencing of tumor-suppressing genes via abnormal levels of oncogenic miRNAs and activation of oncogenes due to lesser concentration of tumor-suppressing miRNAs is the major phenomenon stimulating metastasis. 108 (C) circRNAs control transcription by acting as a sequestering agent for miRNAs and transcription regulatory proteins. 109 (D) Enhanced expression of oncogenic miRNAs by gain-in-function mutations and corresponding silencing of tumor suppressor genes. (E) Sequestering of tumor-suppressing miRNAs by mutated IncRNAs/circRNAs or its down-regulation induced oncogenesis.
List of Some Epigenetic Alterations and Their Mechanism Leading to Cancer.
Epigenetic Biomarkers in Cancer
A biomarker is a biomolecule (present in blood or body tissues) whose detection, measurement, and quantification help to predict normal or pathogenic body conditions. 110 Being accessible, frequent, and stable in body fluids, several epigenetic alterations can act as cancer biomarkers for predictive, diagnostic, and prognostic purposes. 111 Numerous aberrant methylation patterns, NcRNAs, and modifications in histone/chromatin structure have been reported as a potential epigenetic biomarker112–114 for early detection, prognosis, and designing precise and effective therapies. The Encyclopedia of DNA Elements database has achieved the largest amount of epigenomic data from many types of cancerous tissues and 212 cell lines 115 to provide us insight into how the aberrant epigenetic processes shape phenotype in cancer progression. 116 DNA methylation patterns are considered a suitable cancer biomarker to begin accessible in urine, stool, saliva, blood, and tumorous tissues for clinical practices, 117 as shown in Figure 5. The cancer statistics data of 2020 categorized the most usually occurring tumors as breast cancer, succeeded by cancer of the lungs, colorectal, prostate, liver, and stomach. 118 Therefore, the discovery and diagnosis of reliable epigenetic biomarkers, including atypical methylation patterns, histone reshaping, and ncRNAs, are urgently needed. Following are a few biomarkers frequently used for detection and therapeutic responses, as shown in Table 2.
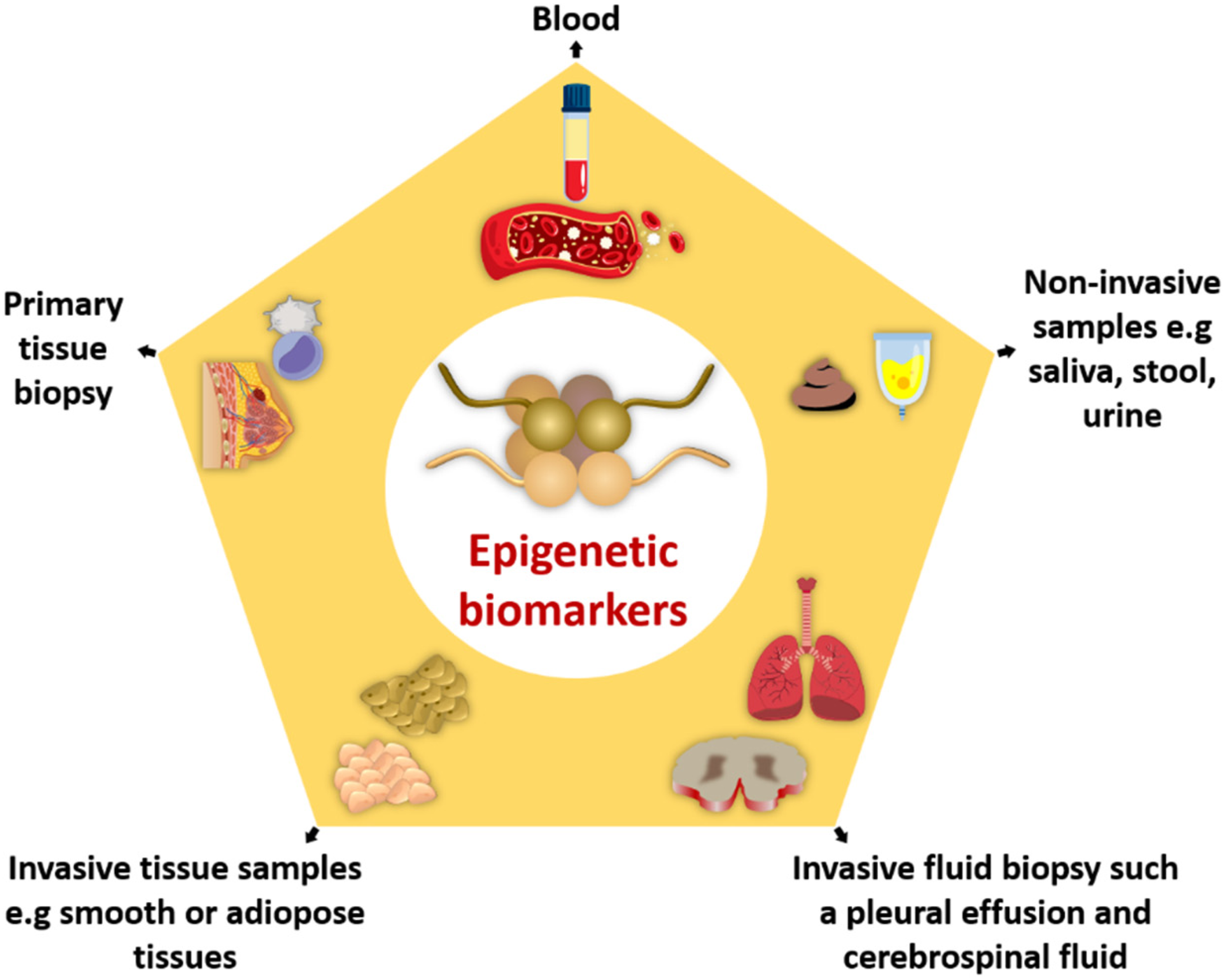
Epigenetic biomarkers in various types of samples.
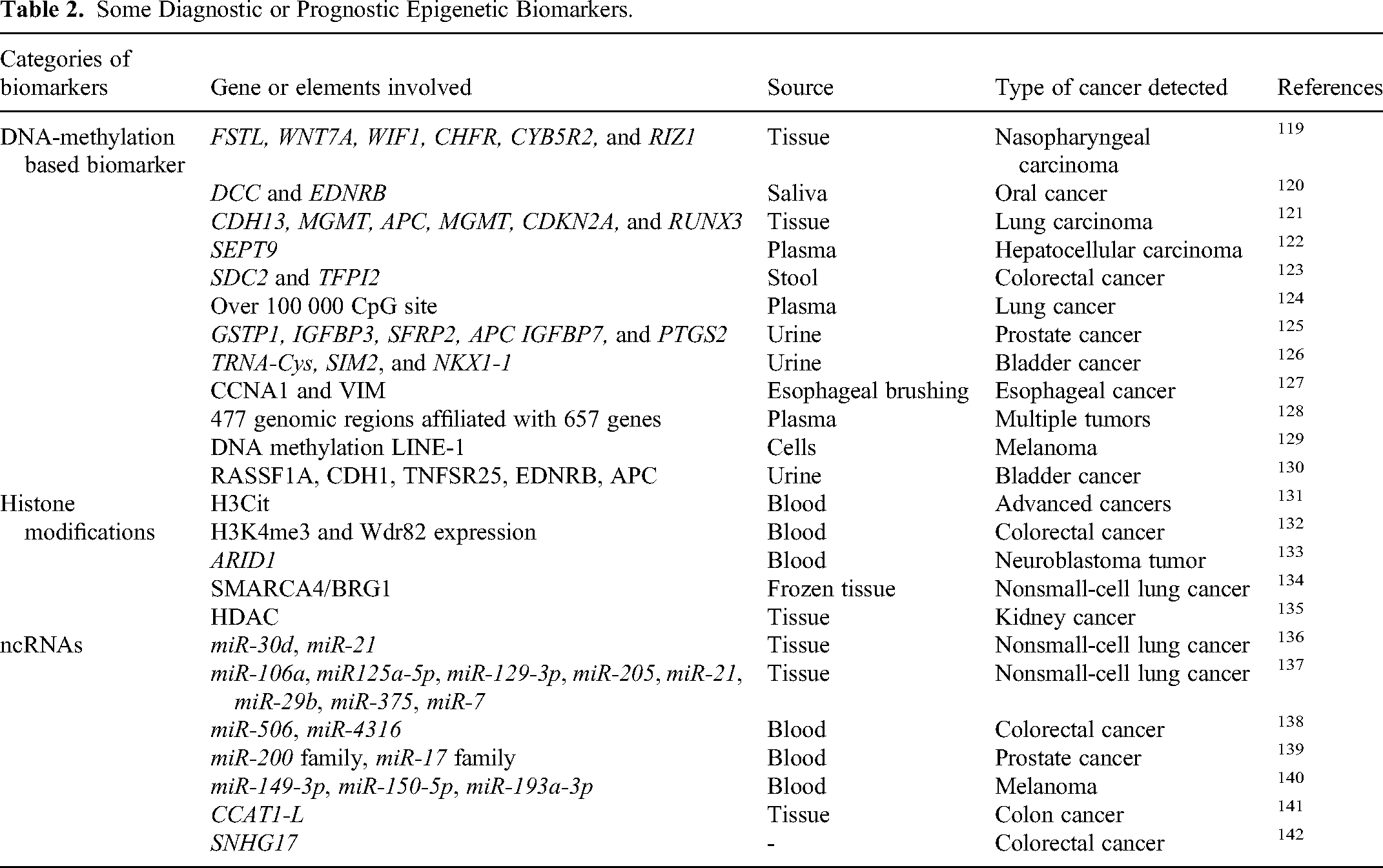
Some Diagnostic or Prognostic Epigenetic Biomarkers.
Epigenetic Therapies for Cancer
Epigenetic alterations have been an attractive therapeutic target because of their detection in the early stages of carcinogenesis. In clinical practices, epigenetic biomarkers are appealing due to their stability in biofluids and tissues. The DNA-based nature of epigenetic biomarkers makes them stable, robust, and technically measurable; for example, DNA methylation can be processed and studied without special management requirements. Epigenetic biomarkers can be detected easily from a lesser amount of biological samples. Unlike protein-based and RNA-based, low-level biomarkers, they can be tracked from entire genomic content (coding or noncoding regions). 143 Current advances in genome-wide sequencing technologies have made significant progress in developing drugs and therapeutic agents for cancer prevention. 49 Inhibitors that obstruct DMTs (DNMTi) and histone deacetylases are the 2 major drugs that are extensively used in cancer treatment. 15 These cytidine analogs inhibit DNMTs by forming covalent complexes with DNA to promote DNMT's degradation or by attaching directly to methylated sites of DNMT. 144 In 2004, the FDA approved nucleoside analogs as the first epigenetic drugs for marketing, named decitabine and azacytidine. 16 Decitabine and azacytidine are considered as a wide range reprograming agents because their dosing in chronic myelomonocytic leukemia and acute myeloid leukemia patients results in the demethylation of repetitive elements, cell growth normalization, and reactivation of tumor suppressors.145, 146 However, the cytotoxic impacts of this drug are significant challenges in therapy, and lower doses are recommended to induce antitumor responses. 147
Histone-deacetylase inhibitor (HDACi) is the other approved category of epigenetic pills that work by decreasing histone acetylation. Romidepsin, vorinostat, belinostat, and panobinostat are approved HDACi that are extensively administered to cure cutaneous T cell lymphoma, peripheral T cell lymphoma, and multiple myeloma. 148 Although a fusion of HDACi and DNMTi has been used to reverse epigenetic aberrations within tumor suppressors and oncogenes, its off-target consequences and scanty sample size have made it controversial for clinical practices. 16 Treatment is further extended to selective therapy targeting specific epigenetic mutations such as tazemetostat (an HMT inhibitor), which is used to treat non-Hodgkin lymphoma. 149 Administering a combination of vorinostat (HDACi) with Panobinostat or Olaparib (PARP inhibitor) enhanced cancer cell apoptosis. It thus exhibited potential anticancer responses in several cancers, including colon adenocarcinoma, breast cancer, and leukemia. 150 Several combinations of epi-drugs, including tazemetostat (EZH2 inhibitors), doxorubicin plus or cyclophosphamide, vincristine, rituximab, prednisolone, and doxorubicin are under clinical evaluation of several carcinomas. 151 Various epi-drug combinations have shown favorable consequences in cancer therapies. A combination of Talacotuzumab, Cytarabine, or chelated zinc, along with DNMT inhibitors, exhibited apoptosis, cell cycle arrest, and growth restriction in patients with severe hematologic malignancies. 152
Epigenetic Research Challenges and Prospects
The role of epigenetics in cancer progression is established and supported by several articles; still, there is a long way to go to apply epigenetics as adaptive potential in cancer initiation. 153 Current research gaps include the development of improved and new technologies and methods for early detection and prevention of cancer by epigenetic modifications. Training of researchers is required for those who are involved in advanced epigenetics methods. The role of epigenetic modifications in changing phenotype and shaping genome function made epigenetic machinery a suitable target for drug development approved by the FDA. Researchers should have a goal of identifying new prognostic biomarkers involved in cancer due to epigenetic changes, commercially available DNA methylation-based assays like Southern hybridization, bisulfite treatment, and nanopore sequencing techniques for detection of modifications and use of epigenetic drugs and inhibitors to eradicate cancer. 154 Future research should deal with the implementation of epigenetic drugs and epigenetic biomarkers in clinical practices through epigenetic therapies and advanced technologies due to the reversible nature of epigenetic alterations. Our knowledge of the development and spread of cancer may be completely transformed using state-of-the-art technologies such as single-cell epigenomics, which may reveal the heterogeneity of epigenetic landscapes inside tumors. 155
Conclusion
Cancer is a progressive disease that breaks all rules of growth and reproduction with the main characteristic of uncontrolled cell division, invading other tissues, and eventually causing host death. It is believed that epigenetic and genetic changes are the reasons behind cancer development. Epigenetic mechanisms like DNA methylation, histone reshaping, and noncoding RNAs are involved in tumor initiation, proliferation, and metastasis. Epigenetic biomarkers are used to identify several kinds of cancer, as they may be treated by different epigenetic therapies, including drugs, inhibitors, RNA-based therapies, and editing technologies, thus contributing to future research. The developing area of epigenetics in cancer research is continually uncovering complex regulatory pathways that contribute to carcinogenesis. New pathways for treatments and diagnostic tools have been made possible by discovering that epigenetic changes are hallmarks of cancer. Using multiomics techniques might help us better understand the heterogeneity of cancer and how different treatments affect it by revealing the complexity of epigenetic fingerprints in various cancer types and providing prospective targets for precision medicine.
Footnotes
Abbreviations
Acknowledgments
This work is carried out with the help of prestigious material from the institute's libraries.
Compliance With Ethical Standards/Ethical Statement
The authors declare that they have no competing interests. We assure the integrity and quality of our research work. It is also stated that there is no plagiarism in this work, and all points taken from other authors are well cited in the text. This study is entirely independent and impartial.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval Statement
Our study did not require ethical board approval because it did not contain human or animal trials. This research did not involve human participants and animals.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.